

Blockade of Autophagy Prevents the Progression of Hyperuricemic Nephropathy Through Inhibiting NLRP3 Inflammasome-Mediated Pyroptosis
- PMID: 35309342
- PMCID: PMC8924517
- DOI: 10.3389/fimmu.2022.858494
Blockade of Autophagy Prevents the Progression of Hyperuricemic Nephropathy Through Inhibiting NLRP3 Inflammasome-Mediated Pyroptosis
Abstract
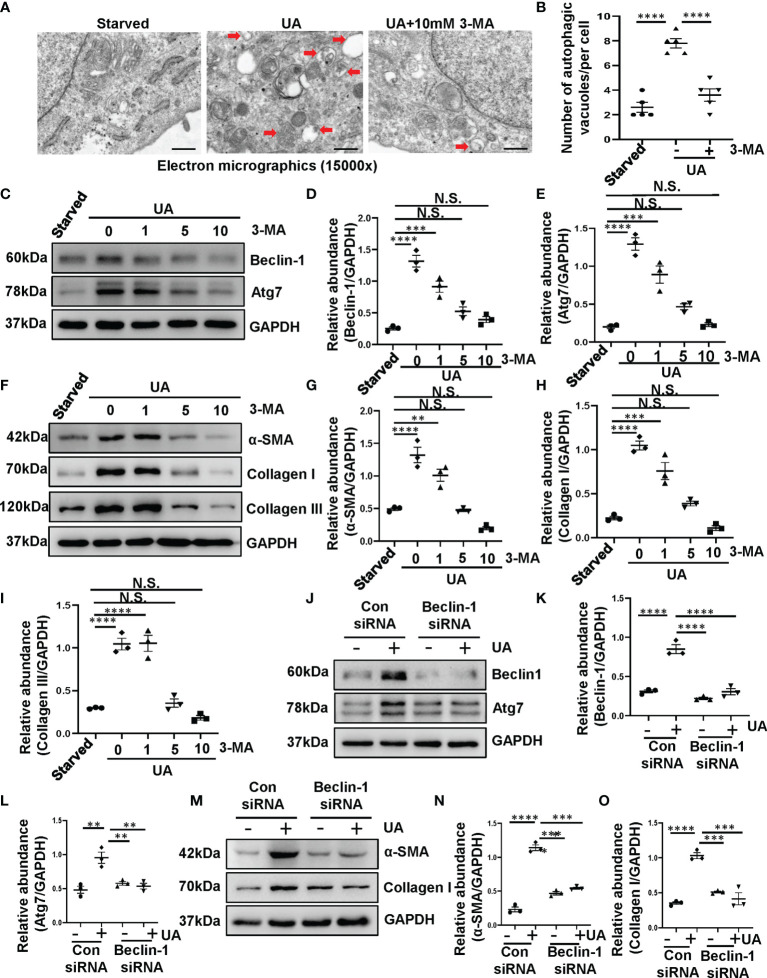
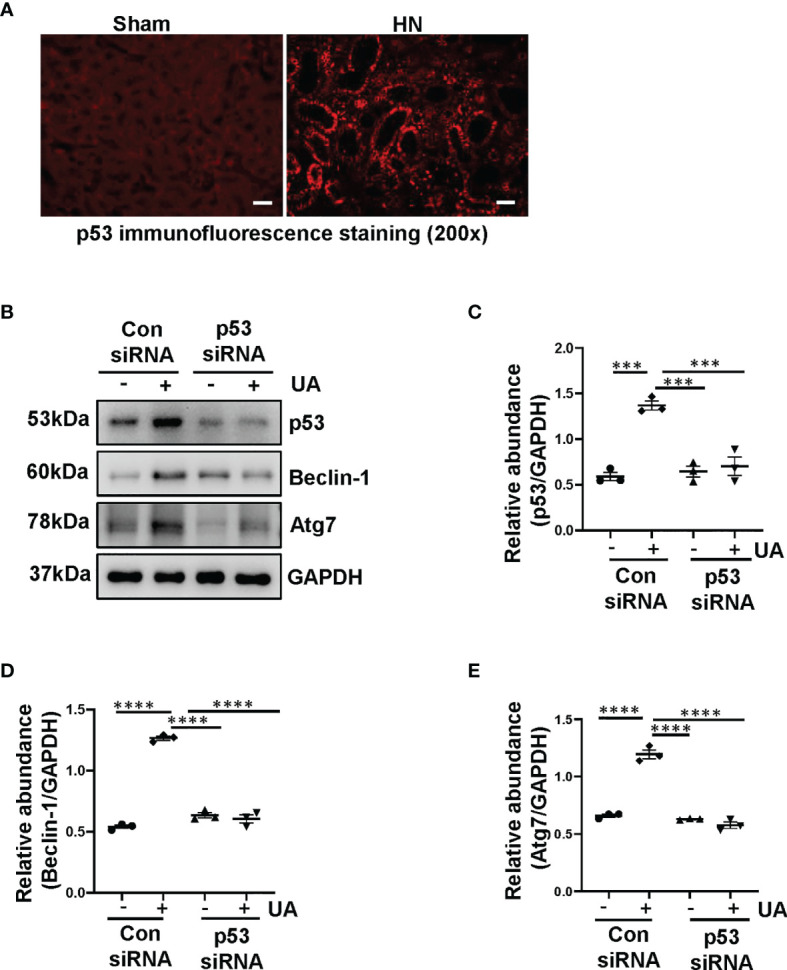
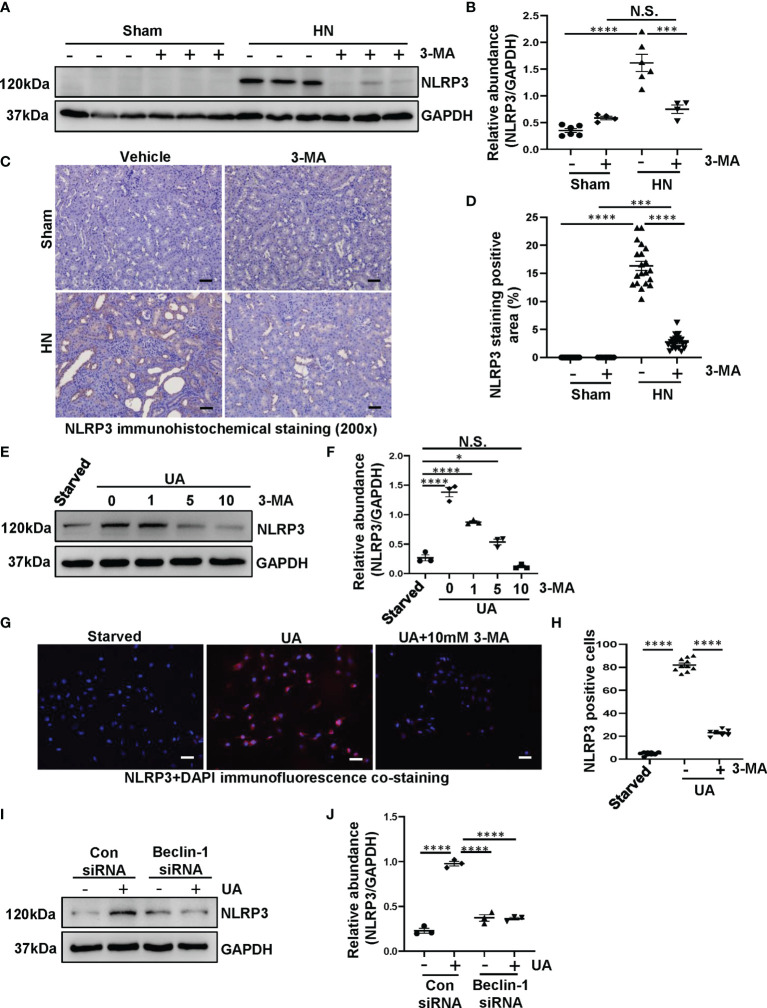
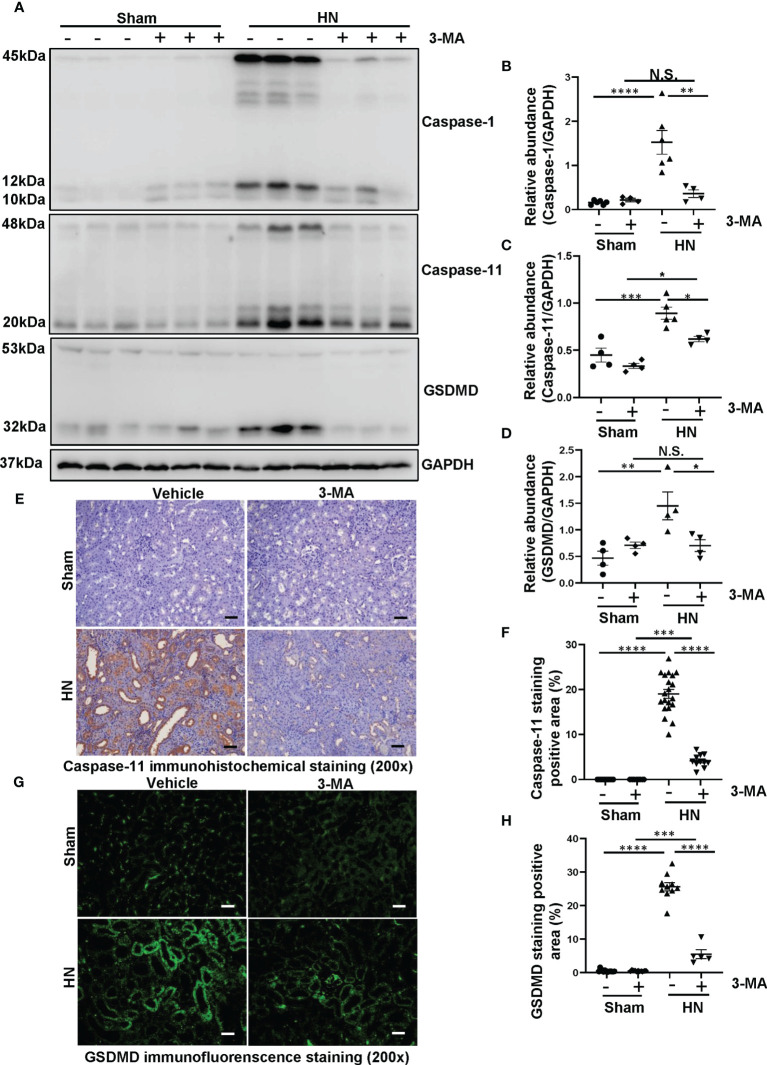
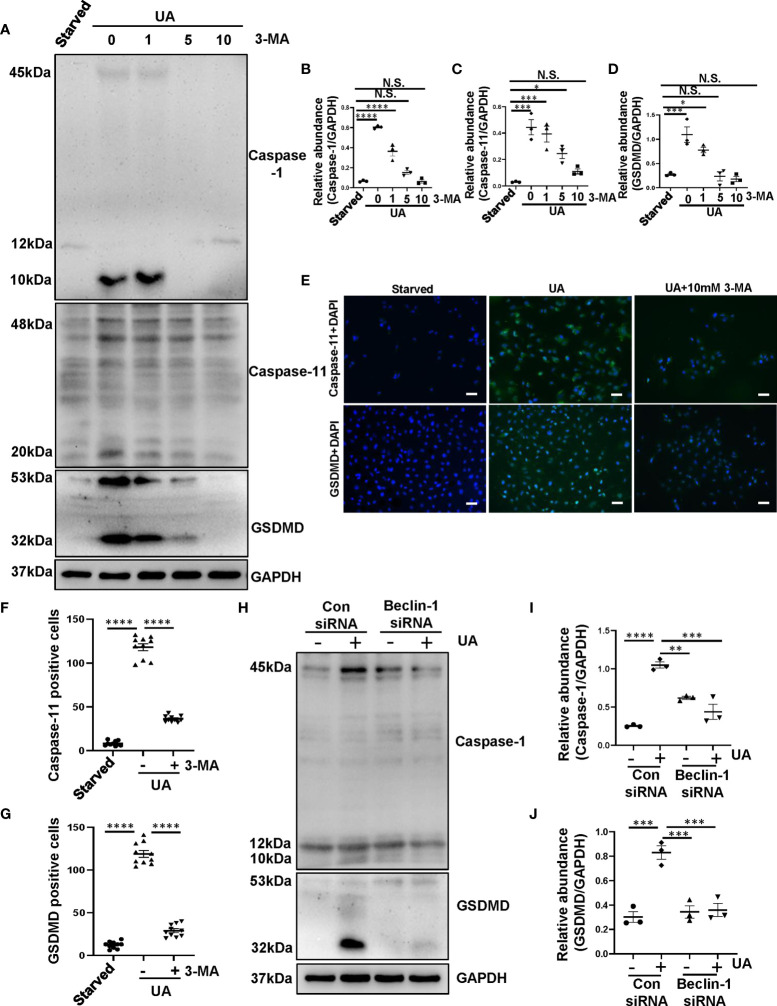
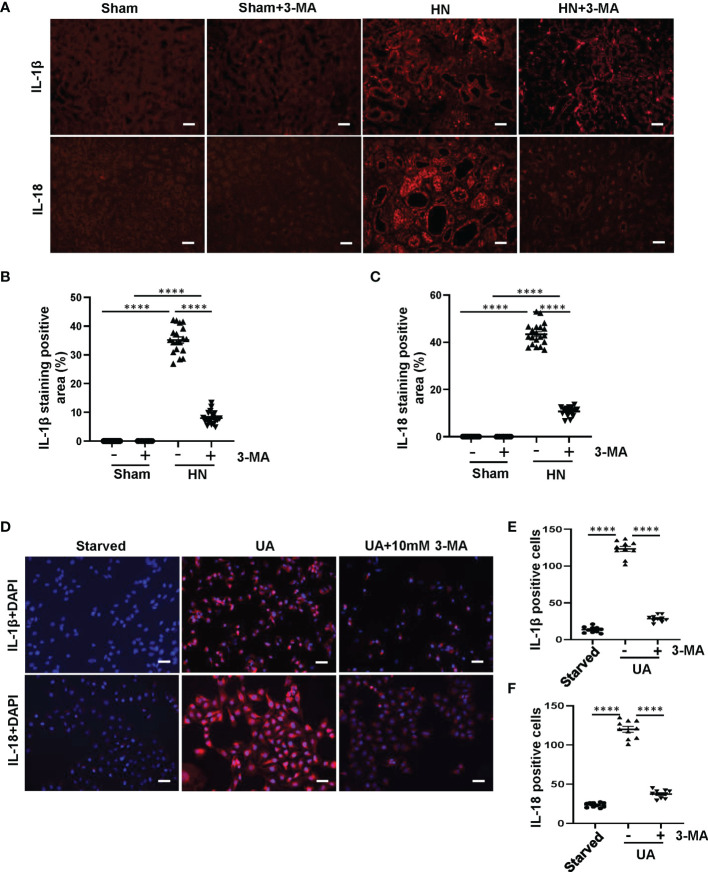
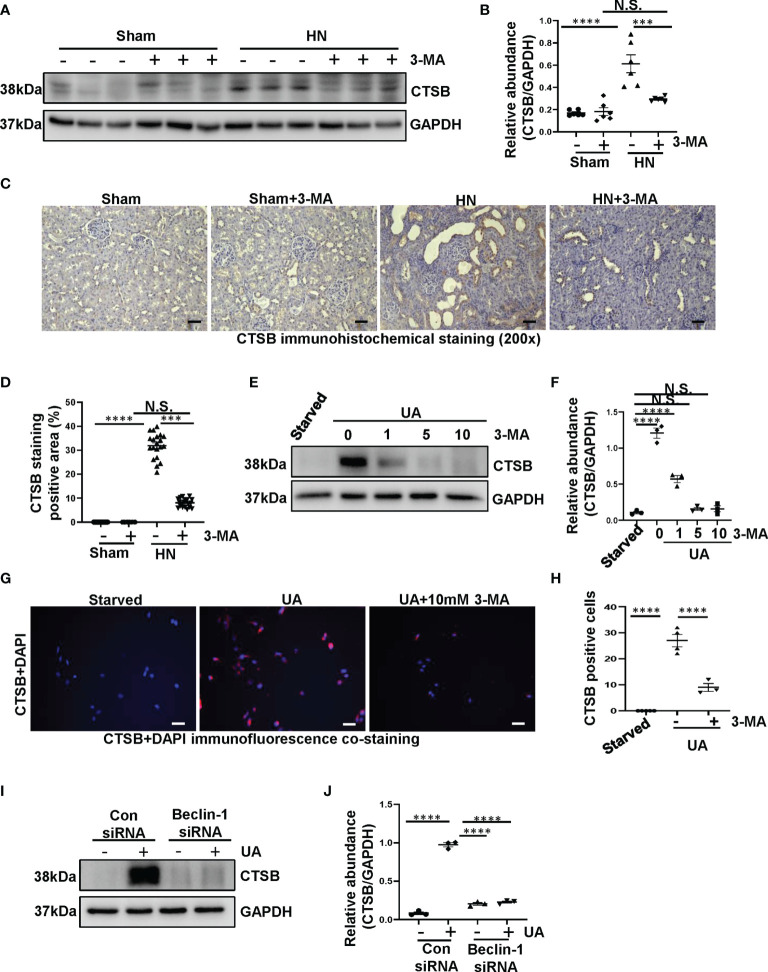
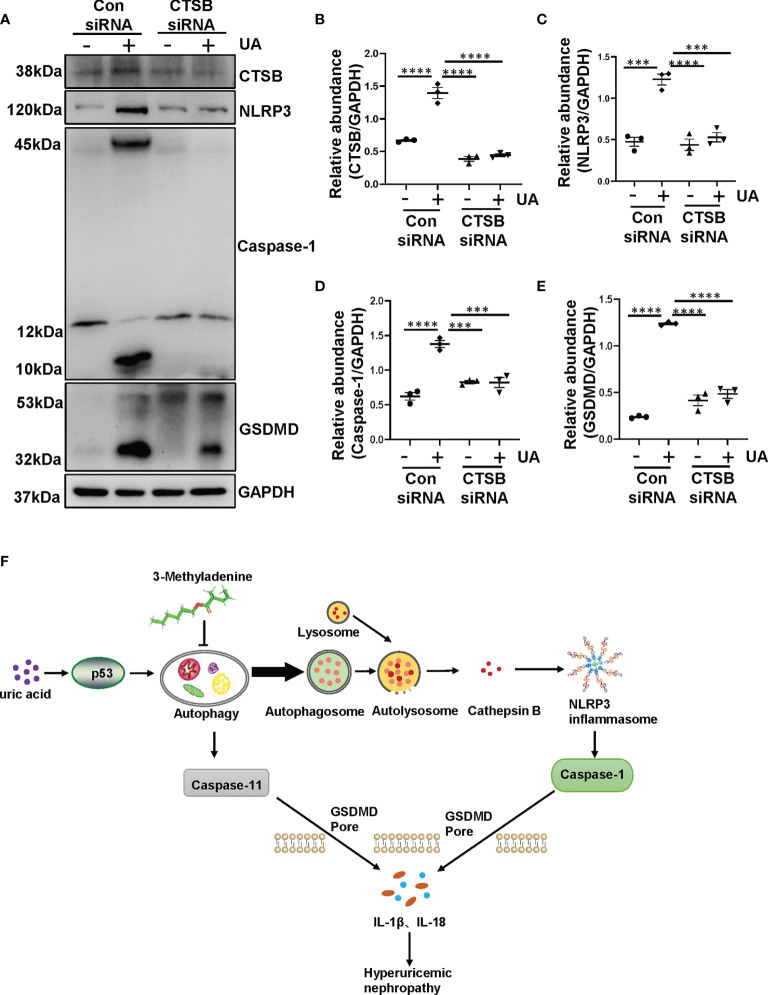
Hyperuricemia has become a common metabolic disease, and is a risk factor for multiple diseases, including chronic kidney disease. Our recent study indicated that following persistent uric acid stimulation, autophagy was activated in rats of hyperuricemic nephropathy (HN) and facilitated the development of renal fibrosis. Nevertheless, the potential mechanism by which autophagy promoted the progression of HN is still not fully elucidated. Thus, in the current study, we investigated the mechanisms of autophagy inhibition on the development of HN. Our data showed that autophagy was activated in human renal tubular cell lines (HK-2) exposure to uric acid. Inhibition of autophagy with 3-methyladenine (3-MA) and transfected with Beclin-1 siRNA prevented uric acid-induced upregulation of α-SMA, Collagen I and Collagen III in HK-2 cells. Moreover, uric acid upregulated autophagy via promoting the p53 pathway. In vivo, we showed that hyperuricemic injury induced the activation of NLRP3 inflammasome and pyroptosis, as evidenced by cleavage of caspase-1 and caspase-11, activation of gasdermin D (GSDMD) and the release of IL-1β and IL-18. Treatment with autophagy inhibitor 3-MA alleviated aforementioned phenomenon. Stimulation with uric acid in HK-2 cells also resulted in NLRP3 inflammasome activation and pyroptotic cell death, however treatment with 3-MA prevented all these responses. Mechanistically, we showed that the elevation of autophagy and degradation of autophagolysosomes resulted in the release of cathepsin B (CTSB), which is related to the activation of NLRP3 inflammasome. CTSB siRNA can inhibit the activation of NLRP3 inflammasome and pyroptosis. Collectively, our results indicate that autophagy inhibition protects against HN through inhibiting NLRP3 inflammasome-mediated pyroptosis. What's more, blockade the release of CTSB plays a crucial role in this process. Thus, inhibition of autophagy may be a promising therapeutic strategy for hyperuricemic nephropathy.
Keywords: NLRP3 inflammasome; autophagy; cathepsin B; hyperuricemic nephropathy; pyroptosis.
Copyright © 2022 Hu, Shi, Chen, Tao, Zhou, Li, Ma, Wang and Liu.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous