

Computational Tools for the Analysis of Uncultivated Phage Genomes
- PMID: 35311574
- PMCID: PMC9199400
- DOI: 10.1128/mmbr.00004-21
Computational Tools for the Analysis of Uncultivated Phage Genomes
Abstract
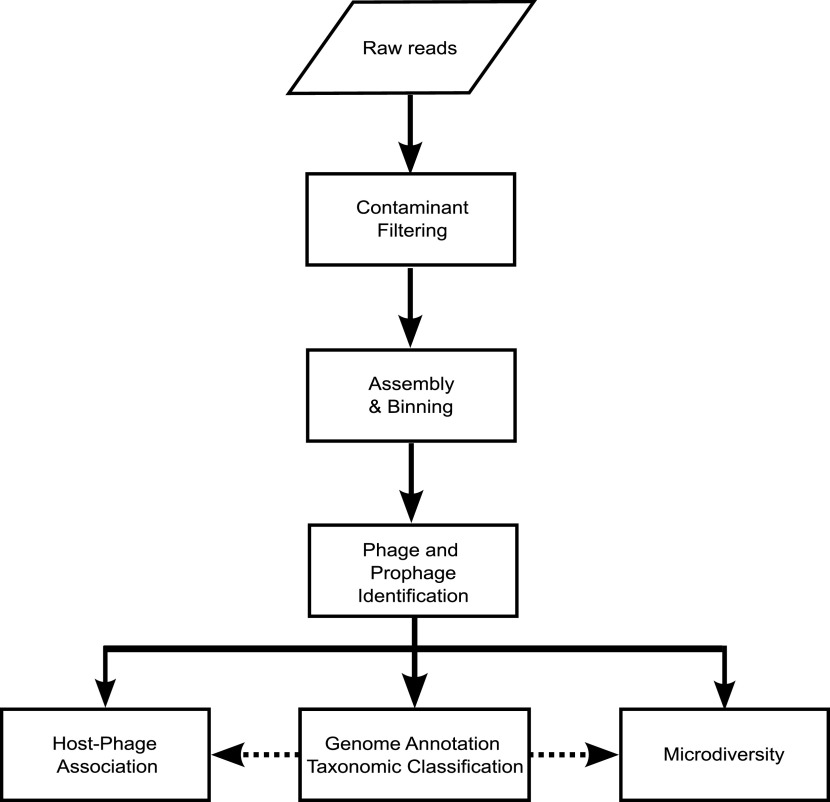
Over a century of bacteriophage research has uncovered a plethora of fundamental aspects of their biology, ecology, and evolution. Furthermore, the introduction of community-level studies through metagenomics has revealed unprecedented insights on the impact that phages have on a range of ecological and physiological processes. It was not until the introduction of viral metagenomics that we began to grasp the astonishing breadth of genetic diversity encompassed by phage genomes. Novel phage genomes have been reported from a diverse range of biomes at an increasing rate, which has prompted the development of computational tools that support the multilevel characterization of these novel phages based solely on their genome sequences. The impact of these technologies has been so large that, together with MAGs (Metagenomic Assembled Genomes), we now have UViGs (Uncultivated Viral Genomes), which are now officially recognized by the International Committee for the Taxonomy of Viruses (ICTV), and new taxonomic groups can now be created based exclusively on genomic sequence information. Even though the available tools have immensely contributed to our knowledge of phage diversity and ecology, the ongoing surge in software programs makes it challenging to keep up with them and the purpose each one is designed for. Therefore, in this review, we describe a comprehensive set of currently available computational tools designed for the characterization of phage genome sequences, focusing on five specific analyses: (i) assembly and identification of phage and prophage sequences, (ii) phage genome annotation, (iii) phage taxonomic classification, (iv) phage-host interaction analysis, and (v) phage microdiversity.
Keywords: computational analysis; microdiversity; phage and prophage identification; phage annotation; phage metagenomics; phage taxonomy; phage-host interaction; uncultivated viruses; viromes.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
Similar articles
-
Comparative Analyses of Bacteriophage Genomes.Methods Mol Biol. 2024;2802:427-453. doi: 10.1007/978-1-0716-3838-5_14. Methods Mol Biol. 2024. PMID: 38819567
-
Construction and characterization of DNA libraries from cultured phages and environmental viromes.Appl Environ Microbiol. 2024 Oct 23;90(10):e0117124. doi: 10.1128/aem.01171-24. Epub 2024 Sep 24. Appl Environ Microbiol. 2024. PMID: 39315792 Free PMC article.
-
Classifying the Unclassified: A Phage Classification Method.Viruses. 2019 Feb 24;11(2):195. doi: 10.3390/v11020195. Viruses. 2019. PMID: 30813498 Free PMC article.
-
Computational approaches to predict bacteriophage-host relationships.FEMS Microbiol Rev. 2016 Mar;40(2):258-72. doi: 10.1093/femsre/fuv048. Epub 2015 Dec 9. FEMS Microbiol Rev. 2016. PMID: 26657537 Free PMC article. Review.
-
Phage hunters: Computational strategies for finding phages in large-scale 'omics datasets.Virus Res. 2018 Jan 15;244:110-115. doi: 10.1016/j.virusres.2017.10.019. Epub 2017 Nov 1. Virus Res. 2018. PMID: 29100906 Review.
Cited by
-
Bacteriophages: A Challenge for Antimicrobial Therapy.Microorganisms. 2025 Jan 7;13(1):100. doi: 10.3390/microorganisms13010100. Microorganisms. 2025. PMID: 39858868 Free PMC article. Review.
-
Dual Nature of Bacteriophages: Friends or Foes in Minimally Processed Food Products-A Comprehensive Review.Viruses. 2025 May 29;17(6):778. doi: 10.3390/v17060778. Viruses. 2025. PMID: 40573368 Free PMC article. Review.
-
KEGG tools for classification and analysis of viral proteins.Protein Sci. 2023 Dec;32(12):e4820. doi: 10.1002/pro.4820. Protein Sci. 2023. PMID: 37881892 Free PMC article.
-
Tools and methodology to in silico phage discovery in freshwater environments.Front Microbiol. 2024 May 31;15:1390726. doi: 10.3389/fmicb.2024.1390726. eCollection 2024. Front Microbiol. 2024. PMID: 38881659 Free PMC article. Review.
-
Campylobacter prophage diversity reveals pervasive recombination between prophages from different Campylobacter species.Microbiol Spectr. 2024 Jan 11;12(1):e0279523. doi: 10.1128/spectrum.02795-23. Epub 2023 Dec 13. Microbiol Spectr. 2024. PMID: 38088548 Free PMC article.
References
-
- Simmonds P, Adams MJ, Benkő M, Breitbart M, Brister JR, Carstens EB, Davison AJ, Delwart E, Gorbalenya AE, Harrach B, Hull R, King AMQ, Koonin EV, Krupovic M, Kuhn JH, Lefkowitz EJ, Nibert ML, Orton R, Roossinck MJ, Sabanadzovic S, Sullivan MB, Suttle CA, Tesh RB, van der Vlugt RA, Varsani A, Zerbini FM. 2017. Consensus statement: virus taxonomy in the age of metagenomics. Nat Rev Microbiol 15:161–168. doi: 10.1038/nrmicro.2016.177. - DOI - PubMed
-
- Cook R, Brown N, Redgwell T, Rihtman B, Barnes M, Clokie M, Stekel DJ, Hobman J, Jones MA, Millard A. 2021. INfrastructure for a PHAge REference Database: identification of large-scale biases in the current collection of cultured phage genomes. Phage (New Rochelle) 2:214–223. doi: 10.1089/phage.2021.0007. - DOI - PMC - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
