

Structural insights into recognition of chemokine receptors by Staphylococcus aureus leukotoxins
- PMID: 35311641
- PMCID: PMC9005193
- DOI: 10.7554/eLife.72555
Structural insights into recognition of chemokine receptors by Staphylococcus aureus leukotoxins
Abstract
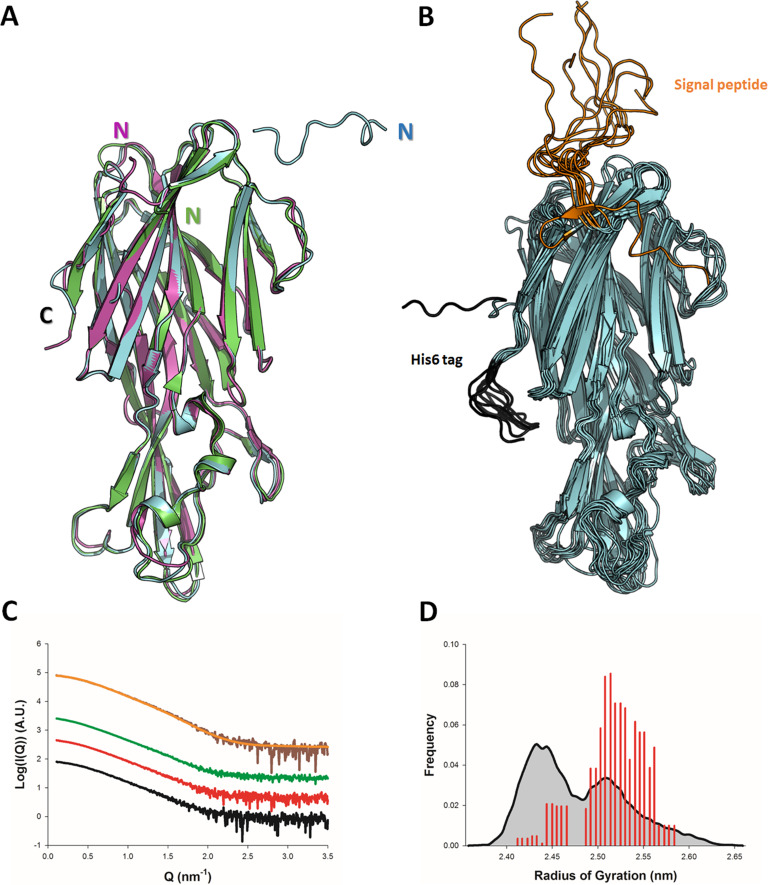
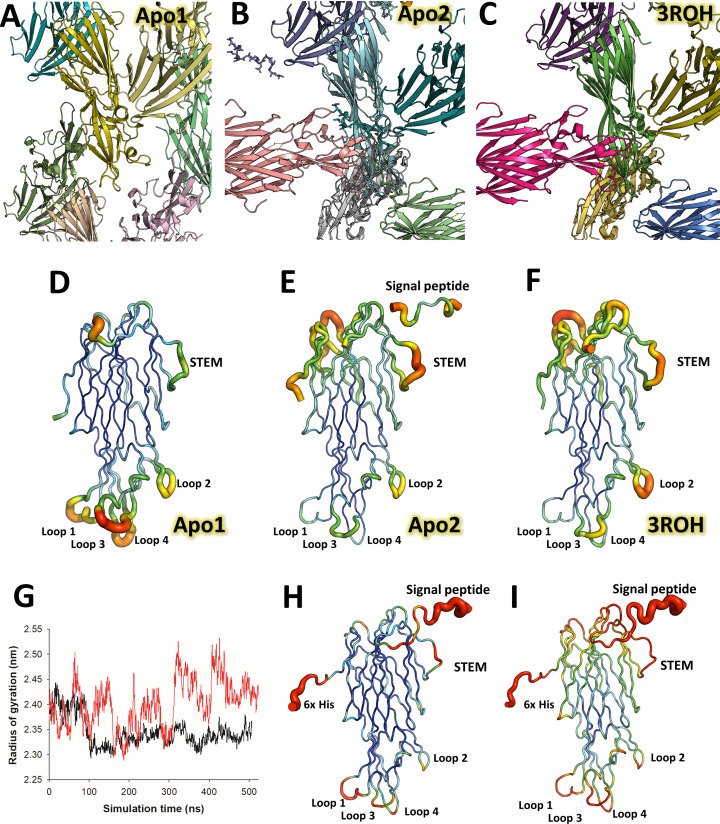
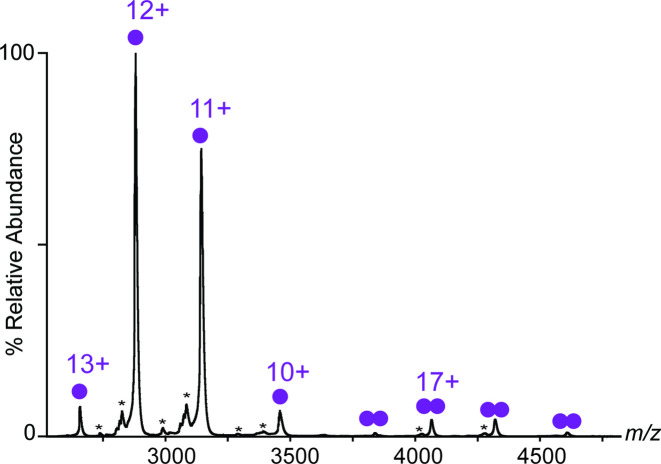
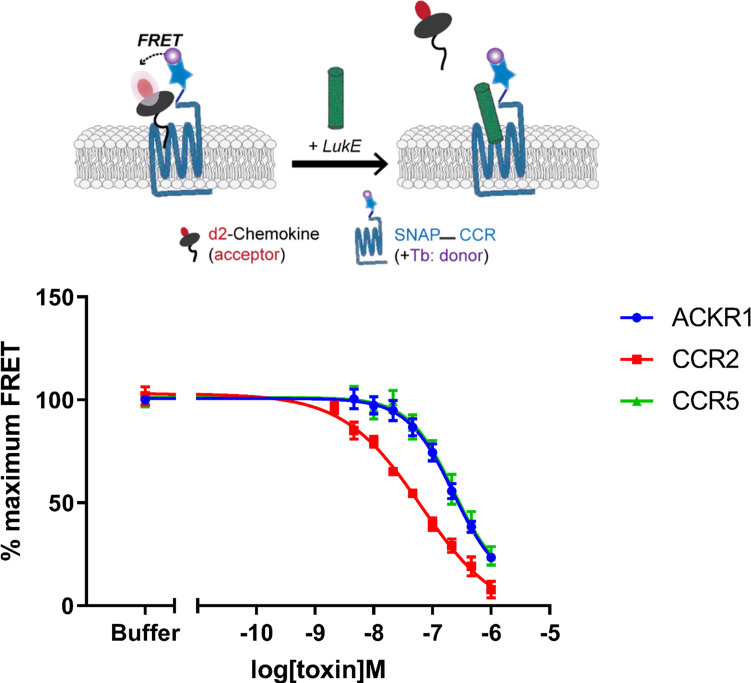
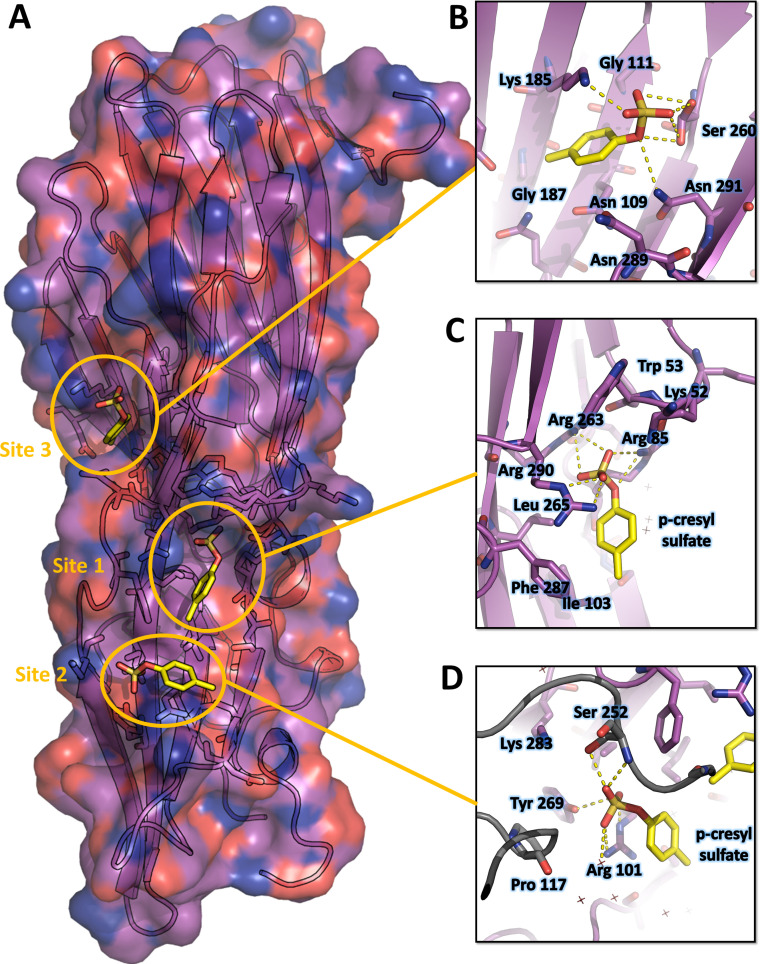
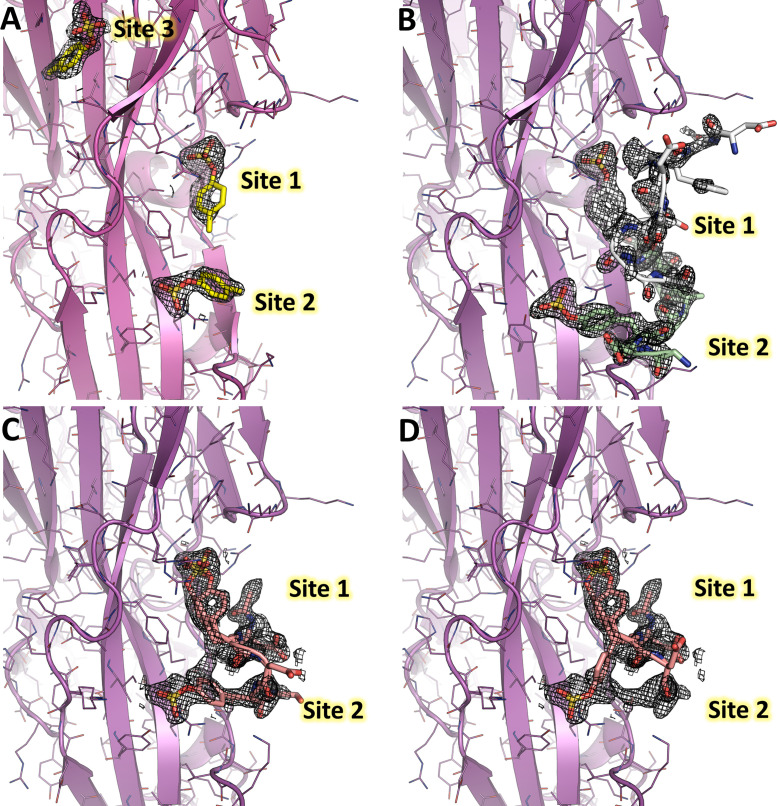
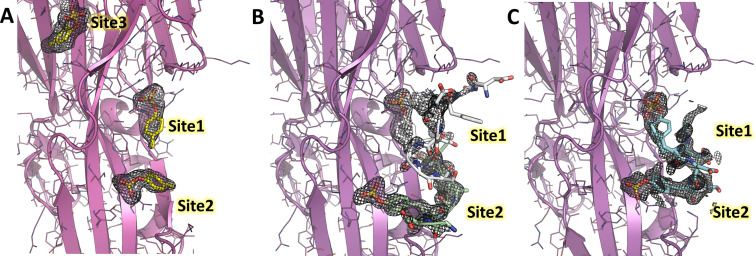
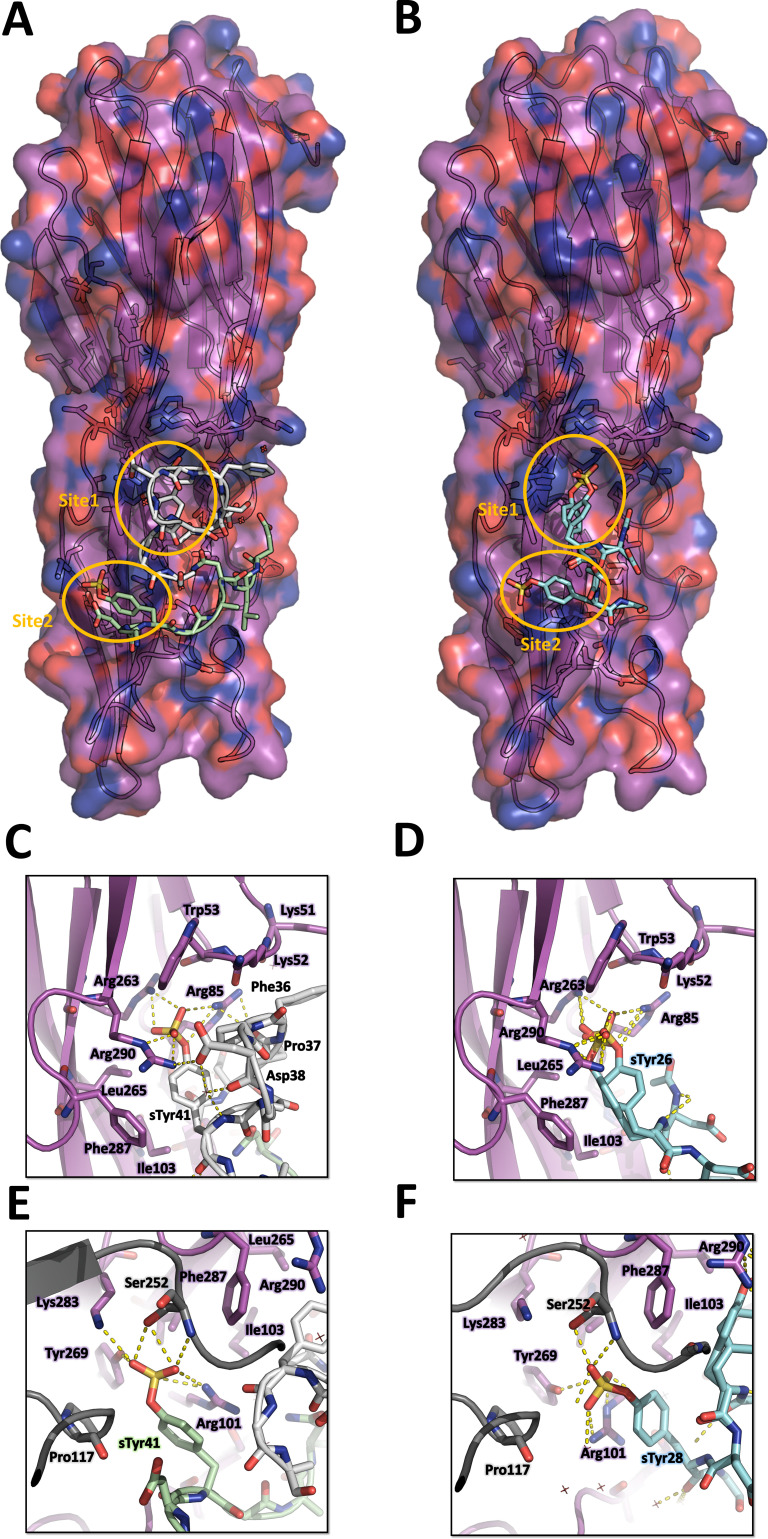
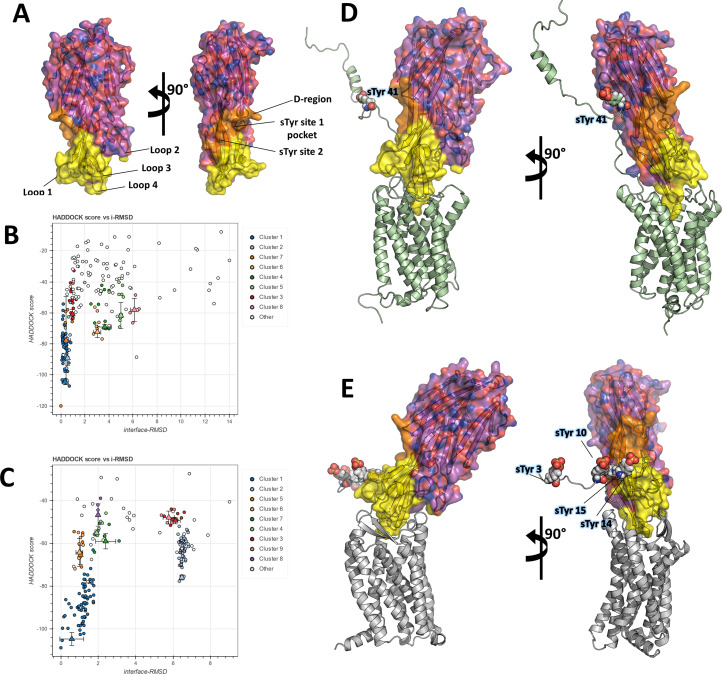
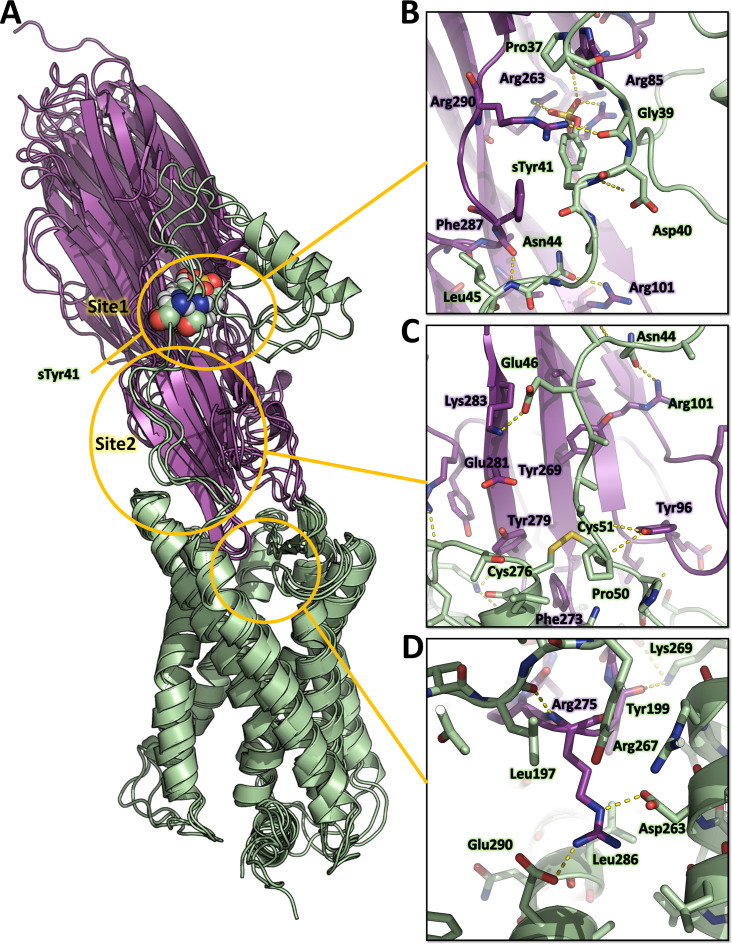
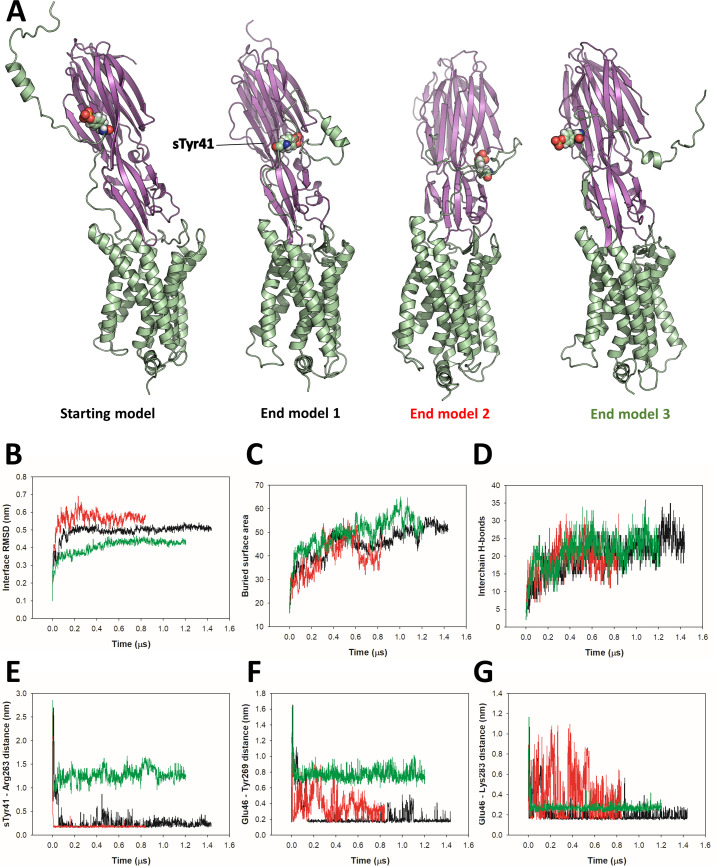
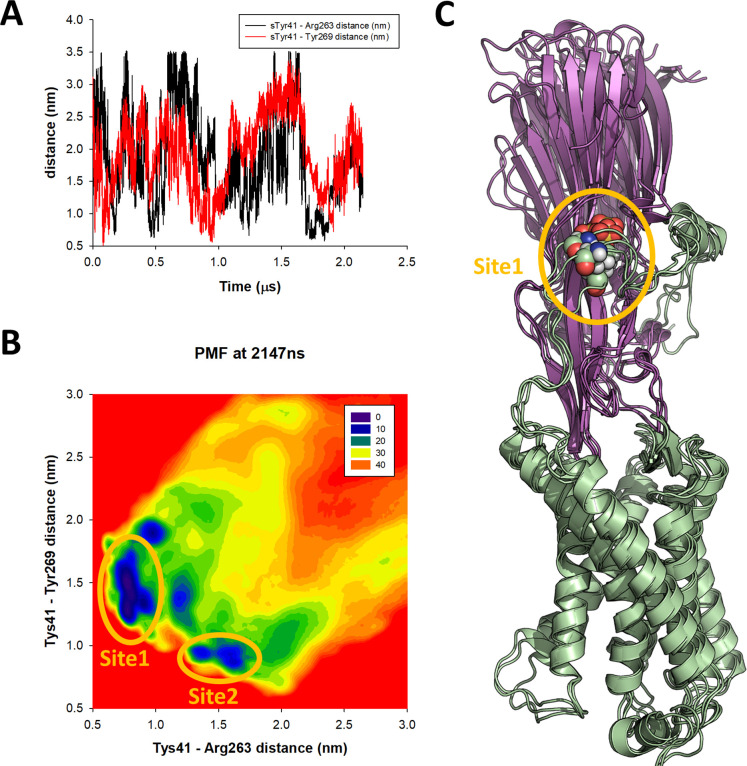
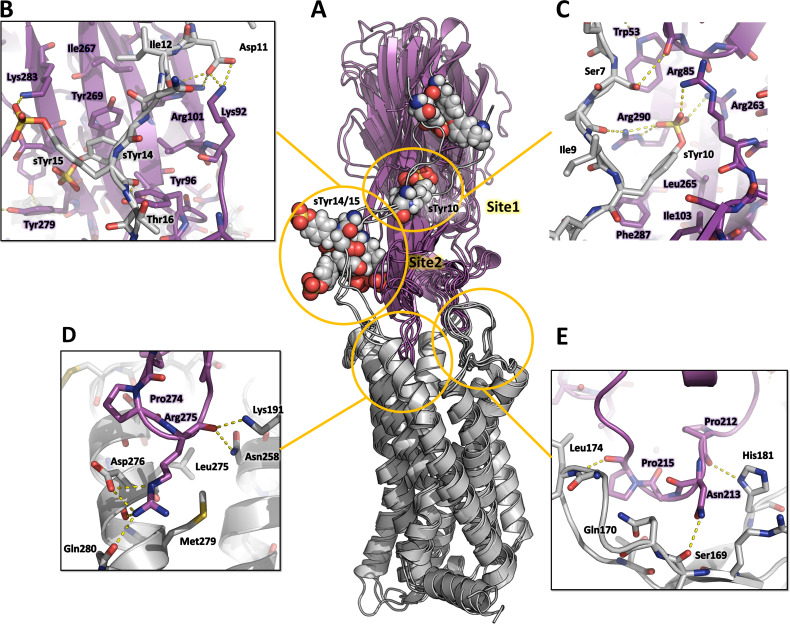
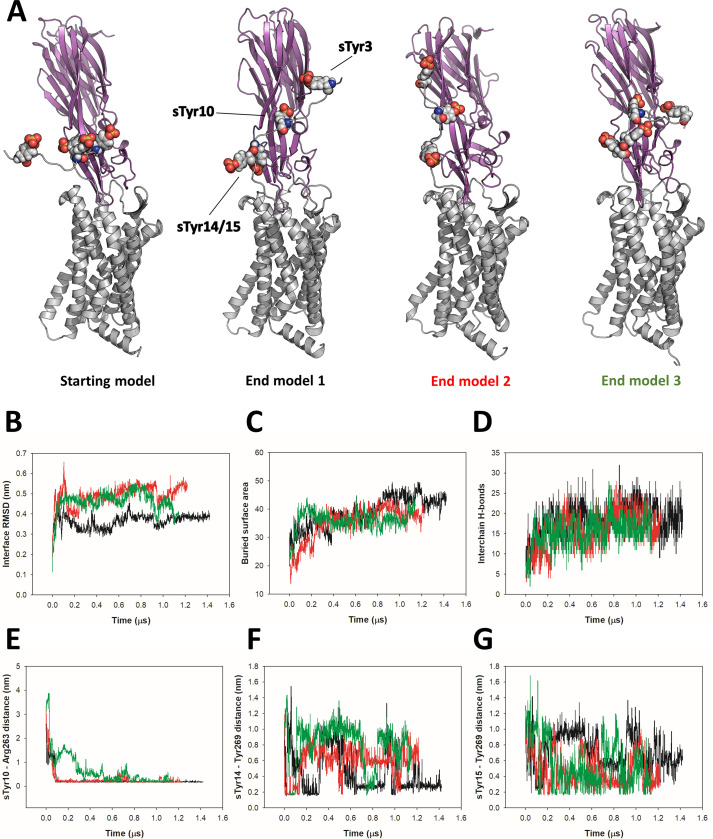
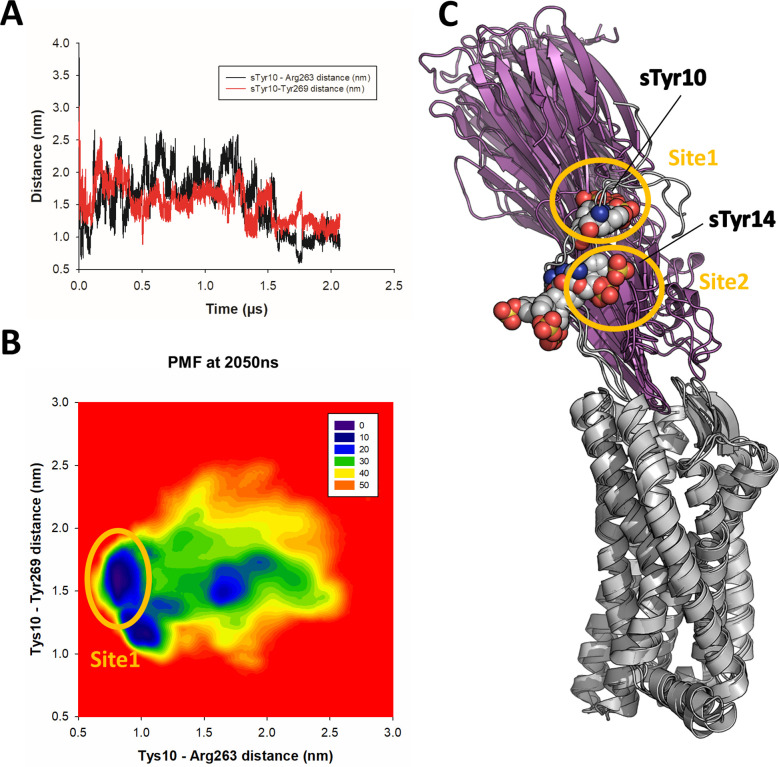
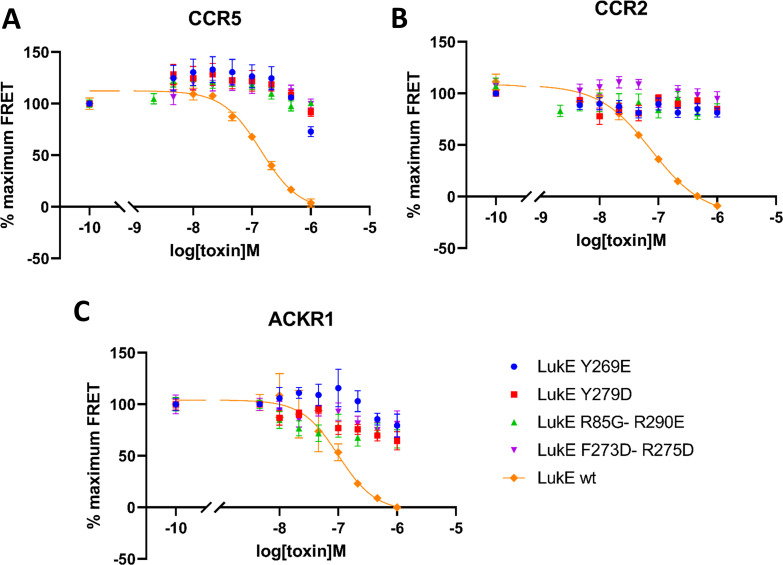
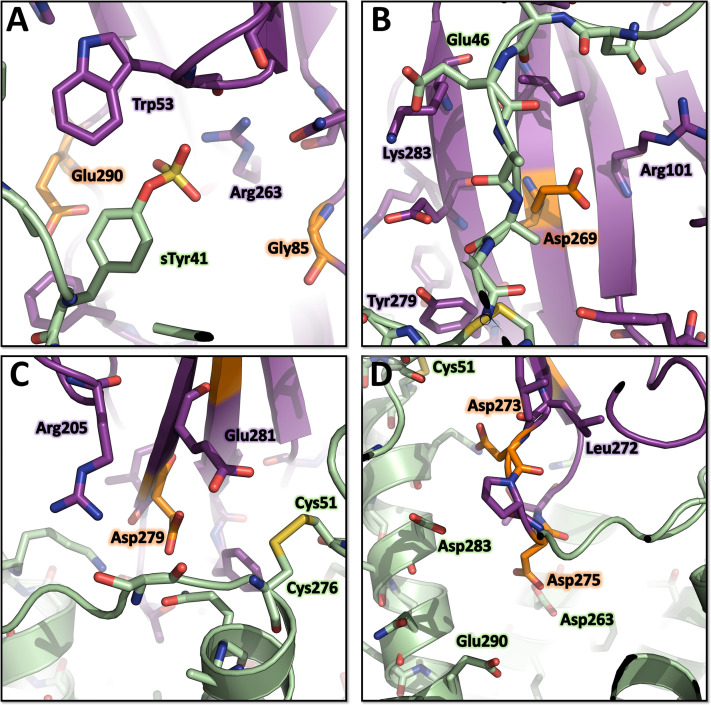
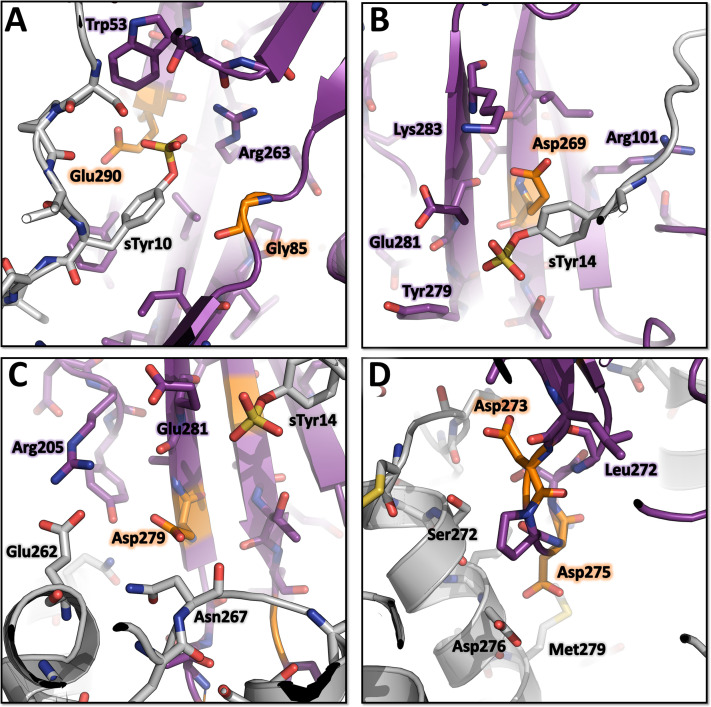
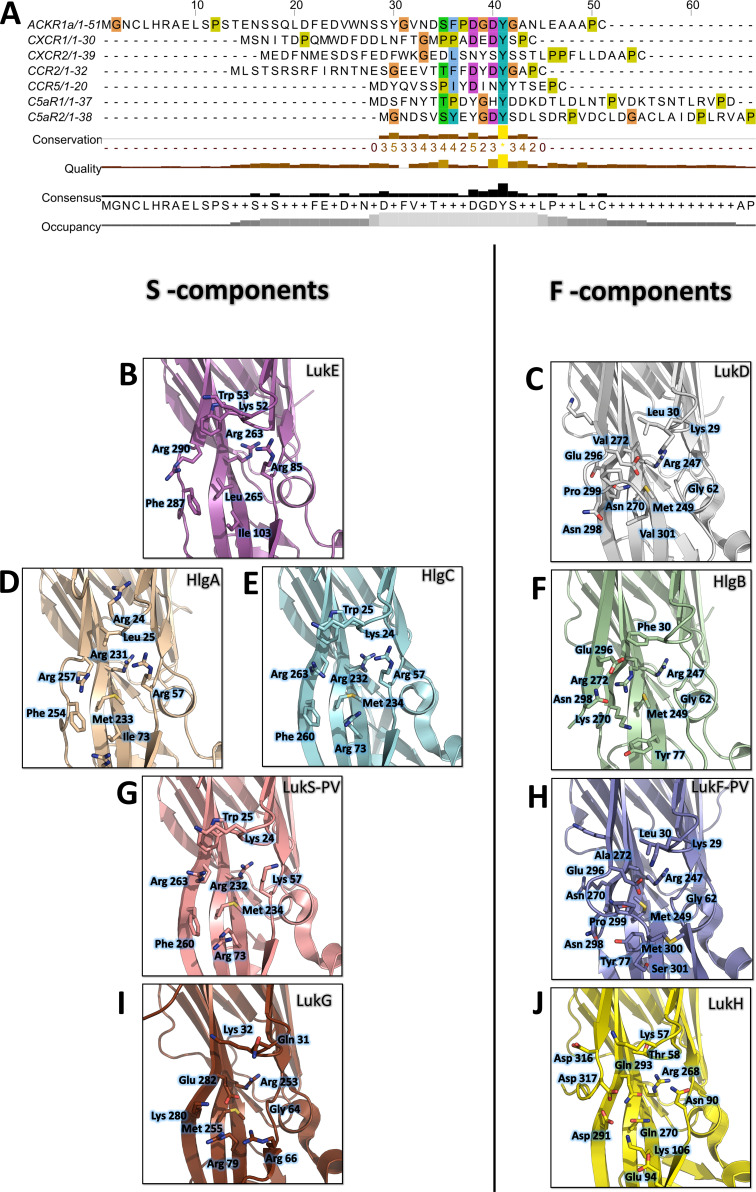
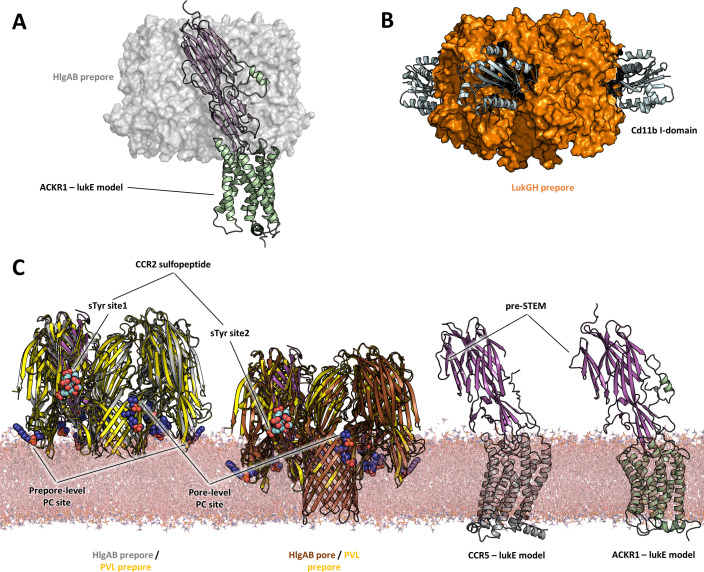
Staphylococcus aureus (SA) leukocidin ED (LukED) belongs to a family of bicomponent pore forming toxins that play important roles in SA immune evasion and nutrient acquisition. LukED targets specific G protein-coupled chemokine receptors to lyse human erythrocytes (red blood cells) and leukocytes (white blood cells). The first recognition step of receptors is critical for specific cell targeting and lysis. The structural and molecular bases for this mechanism are not well understood but could constitute essential information to guide antibiotic development. Here, we characterized the interaction of LukE with chemokine receptors ACKR1, CCR2, and CCR5 using a combination of structural, pharmacological, and computational approaches. First, crystal structures of LukE in complex with a small molecule mimicking sulfotyrosine side chain (p-cresyl sulfate) and with peptides containing sulfotyrosines issued from receptor sequences revealed the location of receptor sulfotyrosine binding sites in the toxins. Then, by combining previous and novel experimental data with protein docking, classical and accelerated weight histogram (AWH) molecular dynamics we propose models of the ACKR1-LukE and CCR5-LukE complexes. This work provides novel insights into chemokine receptor recognition by leukotoxins and suggests that the conserved sulfotyrosine binding pocket could be a target of choice for future drug development.
Keywords: GPCRs; S. aureus; chemokine receptors; crystallography; human; infectious disease; microbiology; molecular biophysics; molecular dynamics; structural biology; sulfotyrosine.
© 2022, Lambey et al.
Conflict of interest statement
PL, OO, XC, FH, LB, PV, CG, FP, SJ, TD, CB, SG, CL No competing interests declared
Figures
Similar articles
-
Staphylococcus aureus Leukocidin LukED and HIV-1 gp120 Target Different Sequence Determinants on CCR5.mBio. 2016 Dec 13;7(6):e02024-16. doi: 10.1128/mBio.02024-16. mBio. 2016. PMID: 27965453 Free PMC article.
-
Host-Receptor Post-Translational Modifications Refine Staphylococcal Leukocidin Cytotoxicity.Toxins (Basel). 2020 Feb 6;12(2):106. doi: 10.3390/toxins12020106. Toxins (Basel). 2020. PMID: 32041354 Free PMC article.
-
Identification of a domain critical for Staphylococcus aureus LukED receptor targeting and lysis of erythrocytes.J Biol Chem. 2020 Dec 11;295(50):17241-17250. doi: 10.1074/jbc.RA120.015757. Epub 2020 Oct 13. J Biol Chem. 2020. PMID: 33051210 Free PMC article.
-
Staphylococcus aureus pore-forming toxins: The interface of pathogen and host complexity.Semin Cell Dev Biol. 2017 Dec;72:101-116. doi: 10.1016/j.semcdb.2017.04.003. Epub 2017 Apr 23. Semin Cell Dev Biol. 2017. PMID: 28445785 Free PMC article. Review.
-
[Pore-forming leukotoxins from Staphylococcus aureus: variability of the target cells and 2 pharmacological processes].Pathol Biol (Paris). 1998 Jun;46(6):435-41. Pathol Biol (Paris). 1998. PMID: 9769878 Review. French.
Cited by
-
CHARMM-GUI Membrane Builder: Past, Current, and Future Developments and Applications.J Chem Theory Comput. 2023 Apr 25;19(8):2161-2185. doi: 10.1021/acs.jctc.2c01246. Epub 2023 Apr 4. J Chem Theory Comput. 2023. PMID: 37014931 Free PMC article. Review.
-
Translocation of linearized full-length proteins through an engineered nanopore under opposing electrophoretic force.Nat Biotechnol. 2024 Aug;42(8):1275-1281. doi: 10.1038/s41587-023-01954-x. Epub 2023 Sep 18. Nat Biotechnol. 2024. PMID: 37723268
-
The chemokine receptor CCR5: multi-faceted hook for HIV-1.Retrovirology. 2024 Jan 23;21(1):2. doi: 10.1186/s12977-024-00634-1. Retrovirology. 2024. PMID: 38263120 Free PMC article. Review.
-
Prospects for targeting ACKR1 in cancer and other diseases.Front Immunol. 2023 Mar 15;14:1111960. doi: 10.3389/fimmu.2023.1111960. eCollection 2023. Front Immunol. 2023. PMID: 37006247 Free PMC article. Review.
-
Gamma-Hemolysin Components: Computational Strategies for LukF-Hlg2 Dimer Reconstruction on a Model Membrane.Int J Mol Sci. 2023 Apr 12;24(8):7113. doi: 10.3390/ijms24087113. Int J Mol Sci. 2023. PMID: 37108277 Free PMC article.
References
-
- Adams PD, Afonine PV, Bunkóczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung L-W, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallographica. Section D, Biological Crystallography. 2010;66:213–221. doi: 10.1107/S0907444909052925. - DOI - PMC - PubMed
-
- Badarau A, Rouha H, Malafa S, Battles MB, Walker L, Nielson N, Dolezilkova I, Teubenbacher A, Banerjee S, Maierhofer B, Weber S, Stulik L, Logan DT, Welin M, Mirkina I, Pleban C, Zauner G, Gross K, Jägerhofer M, Magyarics Z, Nagy E. Context matters: The importance of dimerization-induced conformation of the LukGH leukocidin of Staphylococcus aureus for the generation of neutralizing antibodies. MAbs. 2016;8:1347–1360. doi: 10.1080/19420862.2016.1215791. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
