

Epigenetic Activation of Plasmacytoid DCs Drives IFNAR-Dependent Therapeutic Differentiation of AML
- PMID: 35311997
- PMCID: PMC9355625
- DOI: 10.1158/2159-8290.CD-20-1145
Epigenetic Activation of Plasmacytoid DCs Drives IFNAR-Dependent Therapeutic Differentiation of AML
Abstract
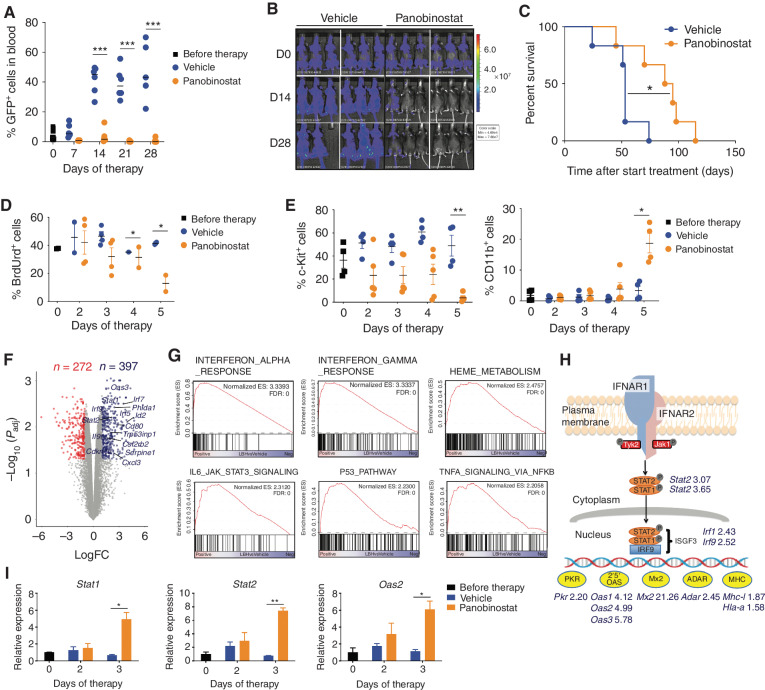
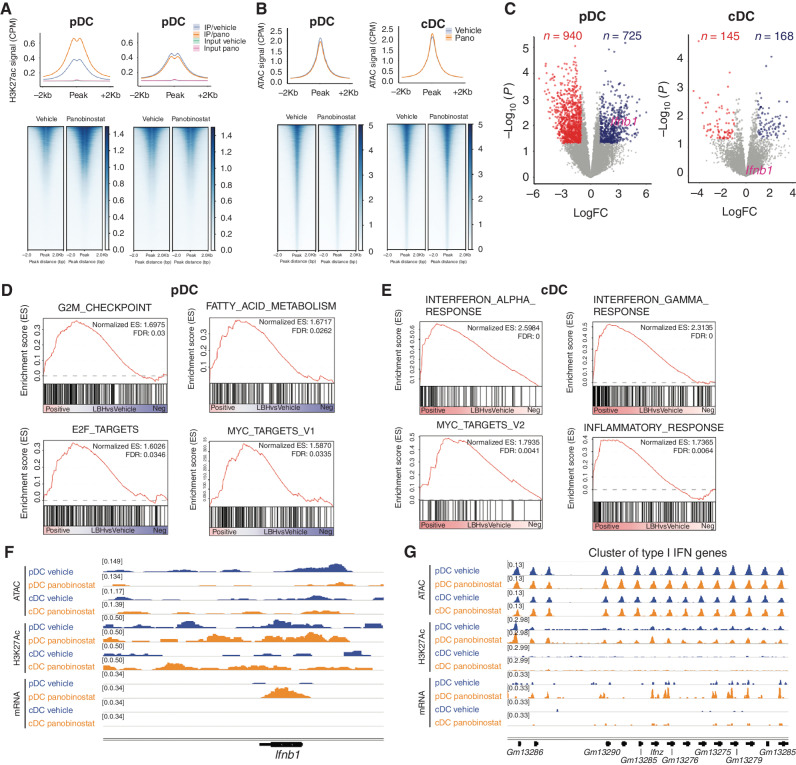
Pharmacologic inhibition of epigenetic enzymes can have therapeutic benefit against hematologic malignancies. In addition to affecting tumor cell growth and proliferation, these epigenetic agents may induce antitumor immunity. Here, we discovered a novel immunoregulatory mechanism through inhibition of histone deacetylases (HDAC). In models of acute myeloid leukemia (AML), leukemia cell differentiation and therapeutic benefit mediated by the HDAC inhibitor (HDACi) panobinostat required activation of the type I interferon (IFN) pathway. Plasmacytoid dendritic cells (pDC) produced type I IFN after panobinostat treatment, through transcriptional activation of IFN genes concomitant with increased H3K27 acetylation at these loci. Depletion of pDCs abrogated panobinostat-mediated induction of type I IFN signaling in leukemia cells and impaired therapeutic efficacy, whereas combined treatment with panobinostat and IFNα improved outcomes in preclinical models. These discoveries offer a new therapeutic approach for AML and demonstrate that epigenetic rewiring of pDCs enhances antitumor immunity, opening the possibility of exploiting this approach for immunotherapies.
Significance: We demonstrate that HDACis induce terminal differentiation of AML through epigenetic remodeling of pDCs, resulting in production of type I IFN that is important for the therapeutic effects of HDACis. The study demonstrates the important functional interplay between the immune system and leukemias in response to HDAC inhibition. This article is highlighted in the In This Issue feature, p. 1397.
©2022 The Authors; Published by the American Association for Cancer Research.
Figures





Comment in
- Cancer Discov. 12:1397.
- Cancer Discov. 12:1397.
References
-
- Ustun C, Marcucci G. Emerging diagnostic and therapeutic approaches in core binding factor acute myeloid leukaemia. Curr Opin Hematol 2015;22:85–91. - PubMed
-
- Solh M, Yohe S, Weisdorf D, Ustun C. Core-binding factor acute myeloid leukemia: heterogeneity, monitoring, and therapy. Am J Hematol 2014;89:1121–31. - PubMed
-
- Frank R, Zhang J, Uchida H, Meyers S, Hiebert SW, Nimer SD. The AML1/ETO fusion protein blocks transactivation of the GM-CSF promoter by AML1B. Oncogene 1995;11:2667–74. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
