

Update from the 5th Edition of the World Health Organization Classification of Head and Neck Tumors: Overview of the 2022 WHO Classification of Head and Neck Neuroendocrine Neoplasms
- PMID: 35312985
- PMCID: PMC9018952
- DOI: 10.1007/s12105-022-01435-8
Update from the 5th Edition of the World Health Organization Classification of Head and Neck Tumors: Overview of the 2022 WHO Classification of Head and Neck Neuroendocrine Neoplasms
Abstract
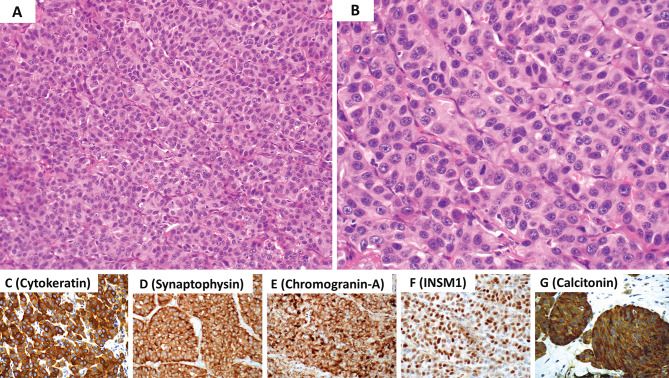
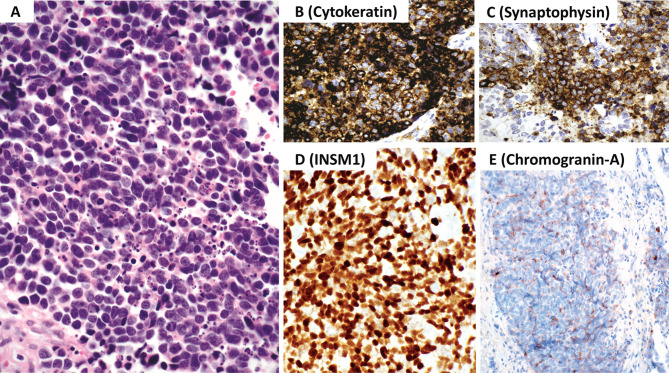
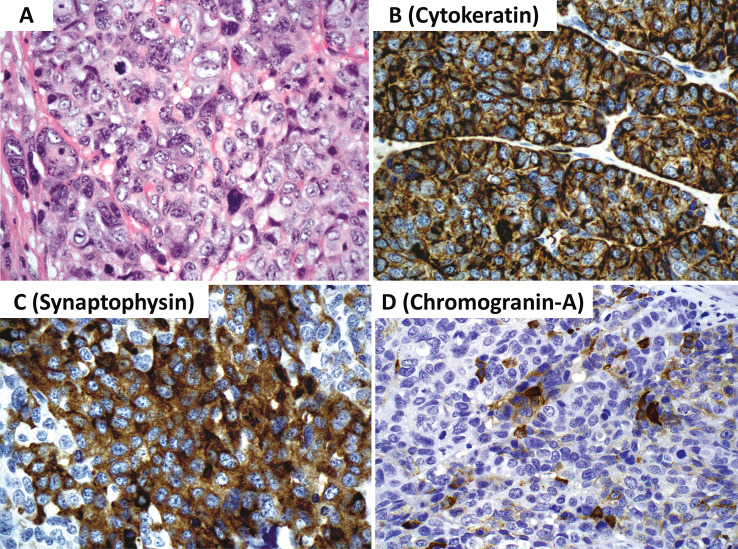
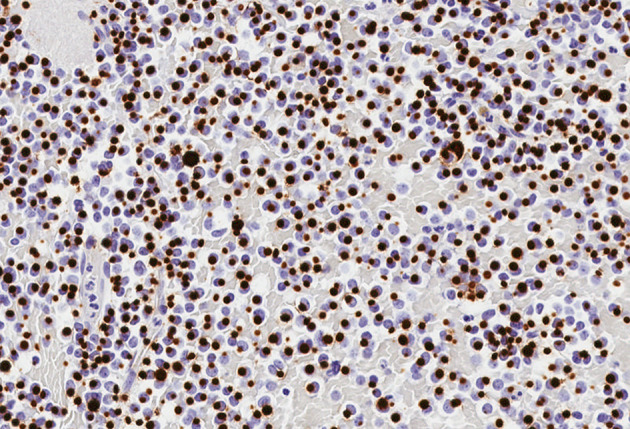
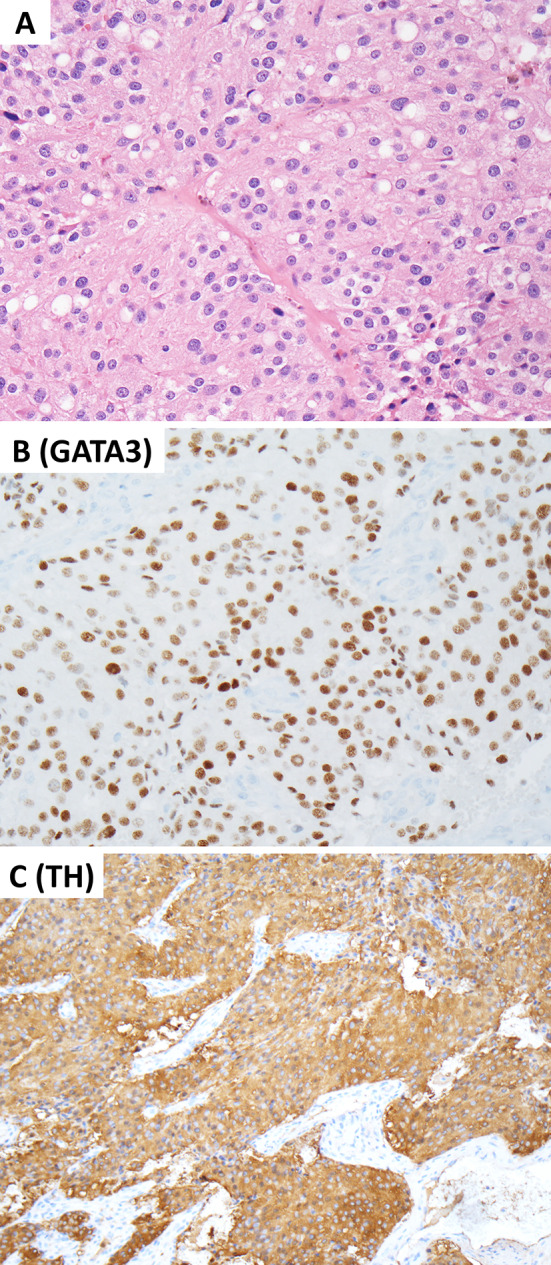
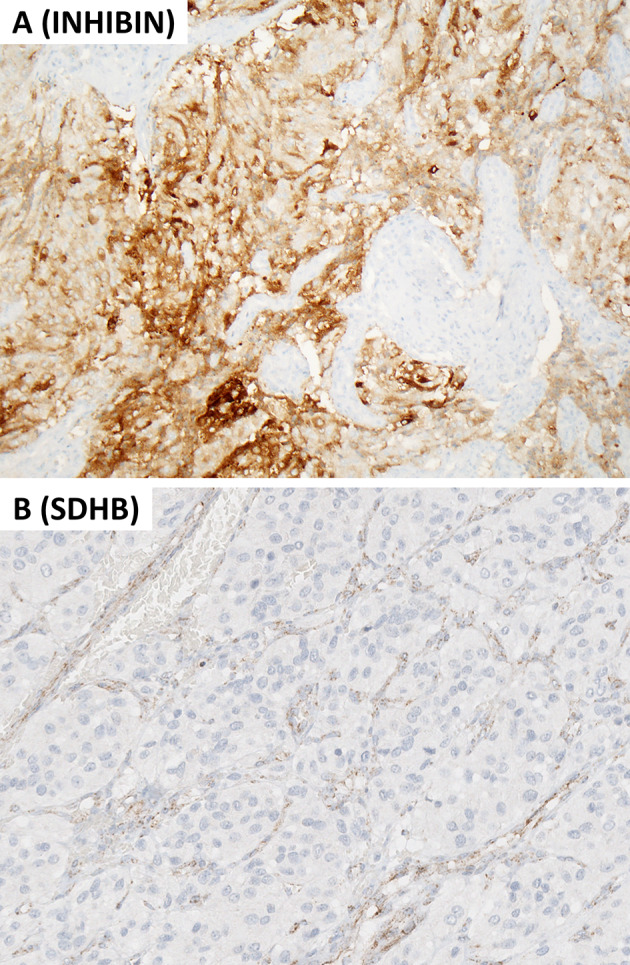
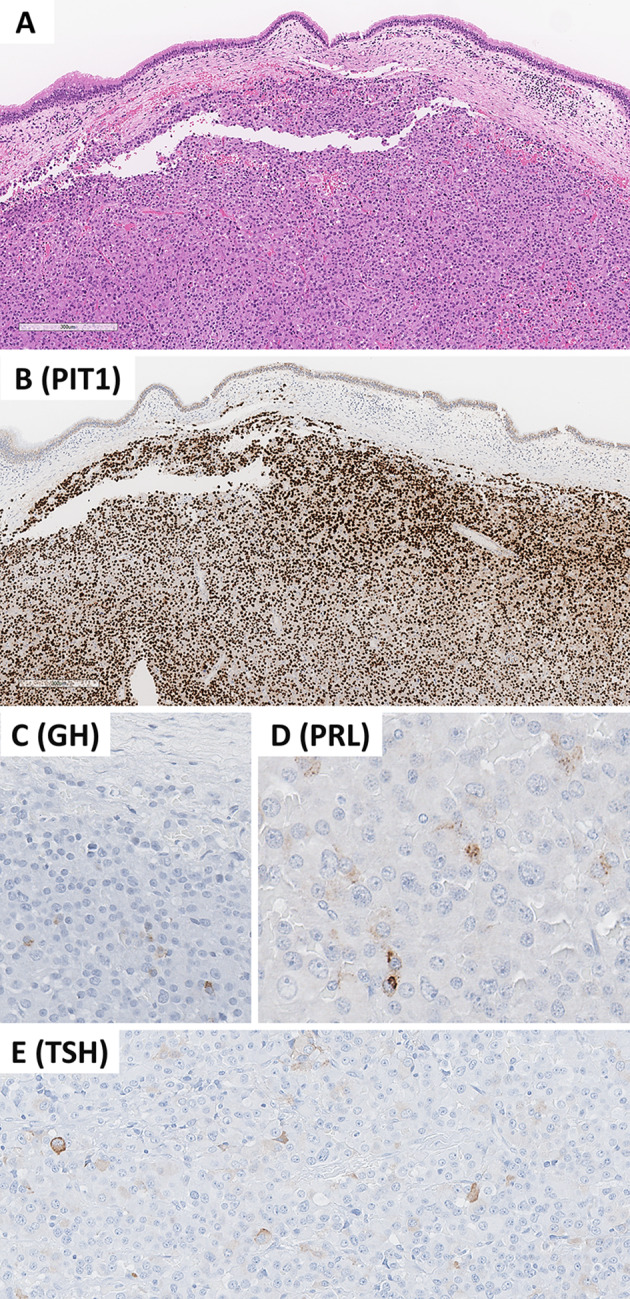
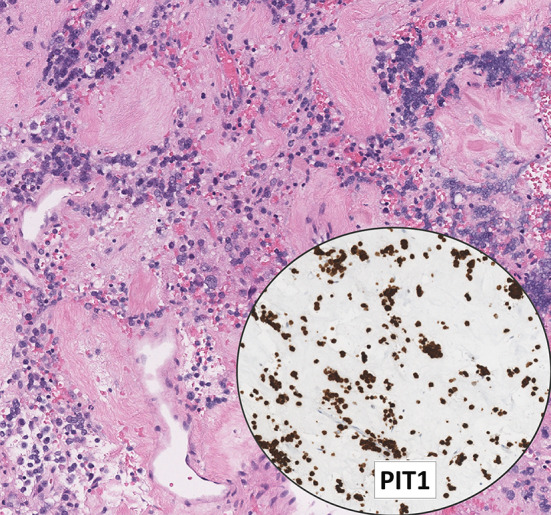
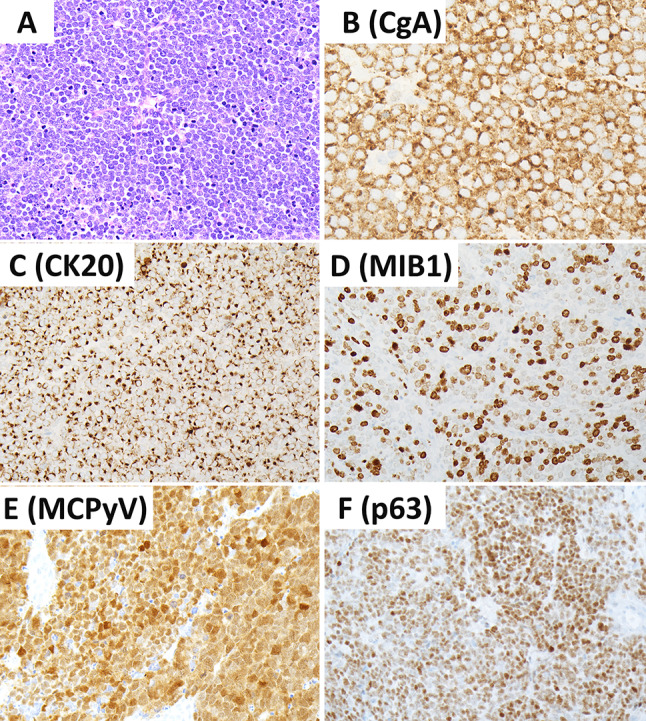
This review article provides a brief overview of the new WHO classification by adopting a question-answer model to highlight the spectrum of head and neck neuroendocrine neoplasms which includes epithelial neuroendocrine neoplasms (neuroendocrine tumors and neuroendocrine carcinomas) arising from upper aerodigestive tract and salivary glands, and special neuroendocrine neoplasms including middle ear neuroendocrine tumors (MeNET), ectopic or invasive pituitary neuroendocrine tumors (PitNET; formerly known as pituitary adenoma) and Merkel cell carcinoma as well as non-epithelial neuroendocrine neoplasms (paragangliomas). The new WHO classification follows the IARC/WHO nomenclature framework and restricts the diagnostic term of neuroendocrine carcinoma to poorly differentiated epithelial neuroendocrine neoplasms. In this classification, well-differentiated epithelial neuroendocrine neoplasms are termed as neuroendocrine tumors (NET), and are graded as G1 NET (no necrosis and < 2 mitoses per 2 mm2; Ki67 < 20%), G2 NET (necrosis or 2-10 mitoses per 2 mm2, and Ki67 < 20%) and G3 NET (> 10 mitoses per 2 mm2 or Ki67 > 20%, and absence of poorly differentiated cytomorphology). Neuroendocrine carcinomas (> 10 mitoses per 2 mm2, Ki67 > 20%, and often associated with a Ki67 > 55%) are further subtyped based on cytomorphological characteristics as small cell and large cell neuroendocrine carcinomas. Unlike neuroendocrine carcinomas, head and neck NETs typically show no aberrant p53 expression or loss of RB reactivity. Ectopic or invasive PitNETs are subtyped using pituitary transcription factors (PIT1, TPIT, SF1, GATA3, ER-alpha), hormones and keratins (e.g., CAM5.2). The new classification emphasizes a strict correlation of morphology and immunohistochemical findings in the accurate diagnosis of neuroendocrine neoplasms. A particular emphasis on the role of biomarkers in the confirmation of the neuroendocrine nature of a neoplasm and in the distinction of various neuroendocrine neoplasms is provided by reviewing ancillary tools that are available to pathologists in the diagnostic workup of head and neck neuroendocrine neoplasms. Furthermore, the role of molecular immunohistochemistry in the diagnostic workup of head and neck paragangliomas is discussed. The unmet needs in the field of head and neck neuroendocrine neoplasms are also discussed in this article. The new WHO classification is an important step forward to ensure accurate diagnosis that will also form the basis of ongoing research in this field.
Keywords: Biomarkers; Head and neck neuroendocrine neoplasms; Merkel cell carcinoma; Neuroendocrine carcinoma; Neuroendocrine tumors; Paraganglioma; Pituitary adenoma; Pituitary neuroendocrine tumor; WHO classification.
© 2022. The Author(s), under exclusive licence to Springer Science+Business Media, LLC, part of Springer Nature.
Conflict of interest statement
The authors declare that this manuscript was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures



References
-
- Rosai J. The origin of neuroendocrine tumors and the neural crest saga. Mod Pathol. 2011;24:S53–S57. - PubMed
-
- Pearse AG. The cytochemistry and ultrastructure of polypeptide hormone-producing cells of the APUD series and the embryologic, physiologic and pathologic implications of the concept. J Histochem Cytochem. 1969;17:303–313. - PubMed
-
- Fontaine J, Le Douarin NM. Analysis of endoderm formation in the avian blastoderm by use of quail-chick chimeras. The problem of the neuroectodermal origin of the cells of the APUD series. J Embryol Exp Morphol. 1977;41:209–222. - PubMed
-
- Oberndorfer S. Karzinoide Tumoren des Dünndarms. Frankf Z Pathol. 1907;1:426–432.
-
- Gosset A, Masson P. Tumeurs endocrines de lappendice. Presse Med. 1914;25:237–240.
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
