

In silico design of a novel nucleotide antiviral agent by free energy perturbation
- PMID: 35313085
- PMCID: PMC9175506
- DOI: 10.1111/cbdd.14042
In silico design of a novel nucleotide antiviral agent by free energy perturbation
Abstract
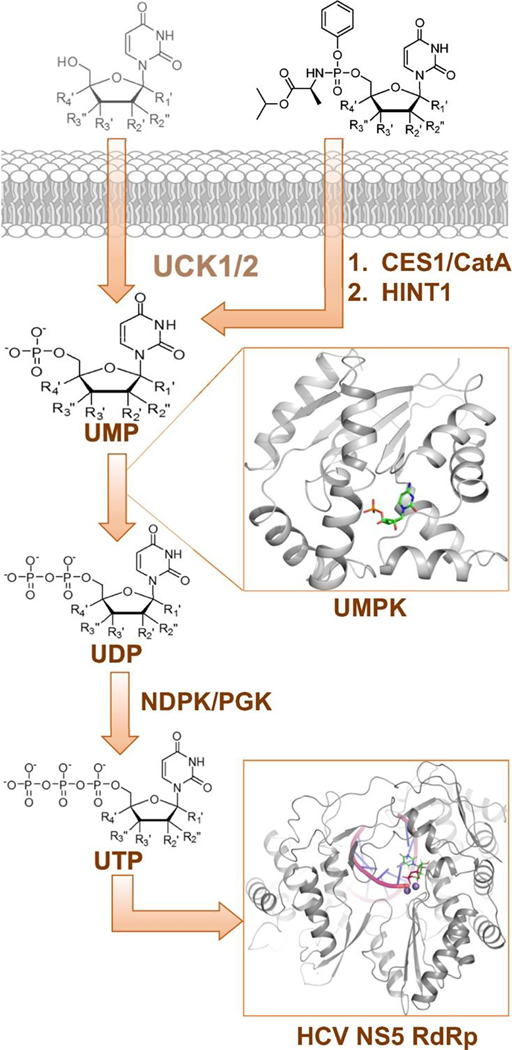
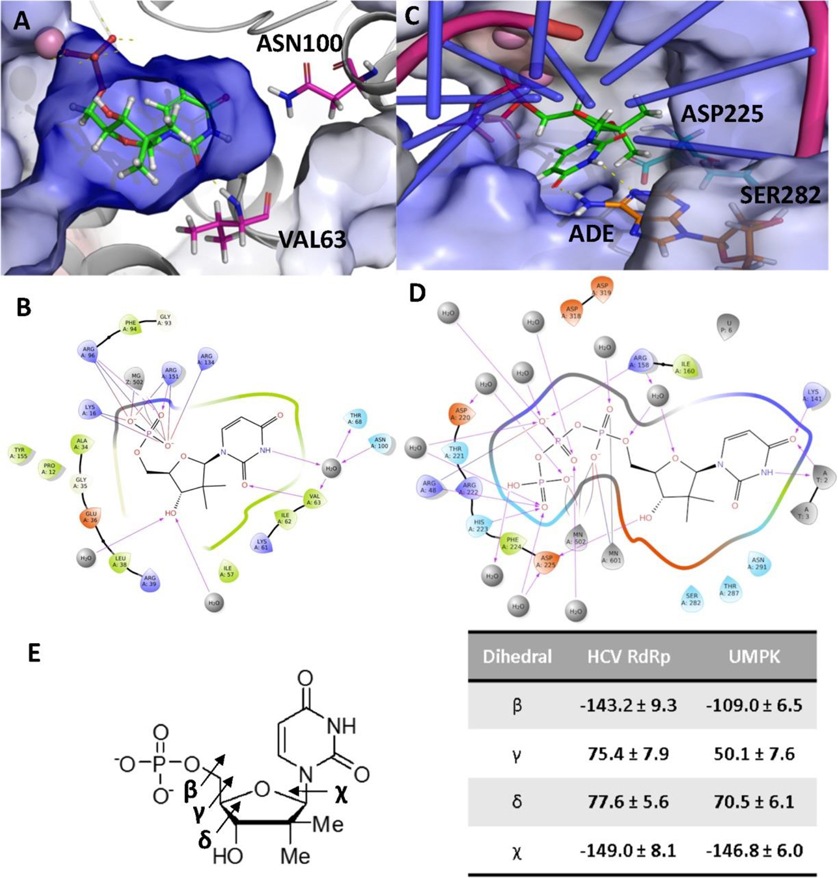
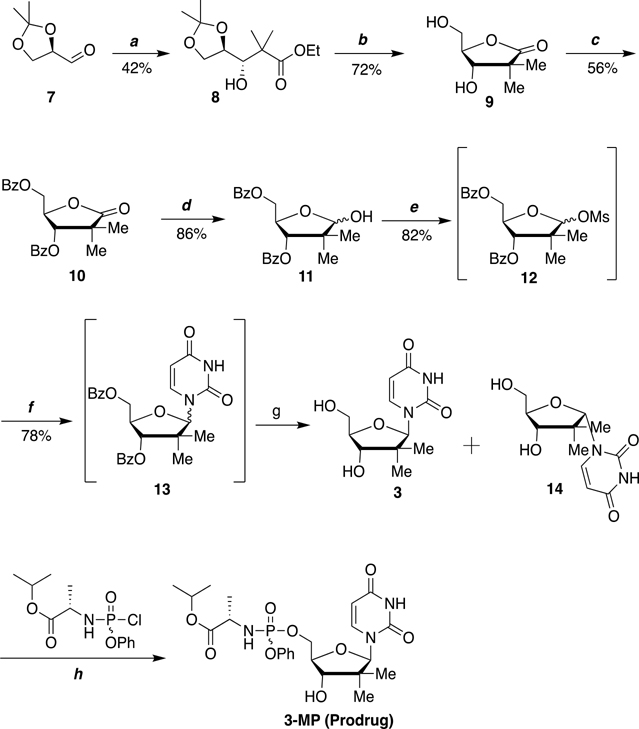
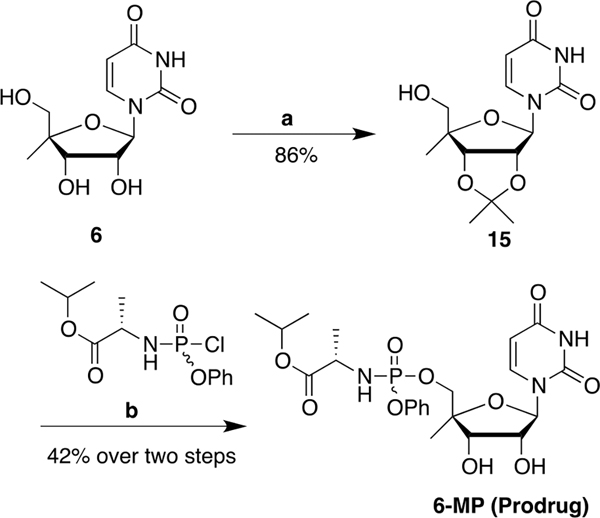
Nucleoside analogs are the backbone of antiviral therapies. Drugs from this class undergo processing by host or viral kinases to form the active nucleoside triphosphate species that selectively inhibits the viral polymerase. It is the central hypothesis that the nucleoside triphosphate analog must be a favorable substrate for the viral polymerase and the nucleoside precursor must be a satisfactory substrate for the host kinases to inhibit viral replication. Herein, free energy perturbation (FEP) was used to predict substrate affinity for both host and viral enzymes. Several uridine 5'-monophosphate prodrug analogs known to inhibit hepatitis C virus (HCV) were utilized in this study to validate the use of FEP. Binding free energies to the host monophosphate kinase and viral RNA-dependent RNA polymerase (RdRp) were calculated for methyl-substituted uridine analogs. The 2'-C-methyl-uridine and 4'-C-methyl-uridine scaffolds delivered favorable substrate binding to the host kinase and HCV RdRp that were consistent with results from cellular antiviral activity in support of our new approach. In a prospective evaluation, FEP results suggest that 2'-C-dimethyl-uridine scaffold delivered favorable monophosphate and triphosphate substrates for both host kinase and HCV RdRp, respectively. Novel 2'-C-dimethyl-uridine monophosphate prodrug was synthesized and exhibited sub-micromolar inhibition of HCV replication. Using this novel approach, we demonstrated for the first time that nucleoside analogs can be rationally designed that meet the multi-target requirements for antiviral activity.
Keywords: RNA-dependent RNA polymerase; alchemical free energy perturbation; flavivirus; nucleoside antiviral agents; structure-based drug design; viral polymerase.
© 2022 John Wiley & Sons A/S.
Figures
References
-
- Alexandre F-R, Badaroux E, Bilello JP, Bot S, Bouisset T, Brandt G, Cappelle S, Chapron C, Chaves D, & Convard T (2017). The discovery of idx21437: Design, synthesis and antiviral evaluation of 2′-α-chloro-2′-β-c-methyl branched uridine pronucleotides as potent liver-targeted hcv polymerase inhibitors. Bioorganic & medicinal chemistry letters, 27(18), 4323–4330. - PubMed
-
- Appleby TC, Perry JK, Murakami E, Barauskas O, Feng J, Cho A, Fox D, Wetmore DR, McGrath ME, & Ray AS (2015). Structural basis for rna replication by the hepatitis c virus polymerase. Science, 347(6223), 771–775. - PubMed
-
- Caglioti C, Lalle E, Castilletti C, Carletti F, Capobianchi MR, & Bordi L (2013). Chikungunya virus infection: An overview. New Microbiol, 36(3), 211–227. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
