

A novel immunopeptidomic-based pipeline for the generation of personalized oncolytic cancer vaccines
- PMID: 35314027
- PMCID: PMC8989416
- DOI: 10.7554/eLife.71156
A novel immunopeptidomic-based pipeline for the generation of personalized oncolytic cancer vaccines
Abstract
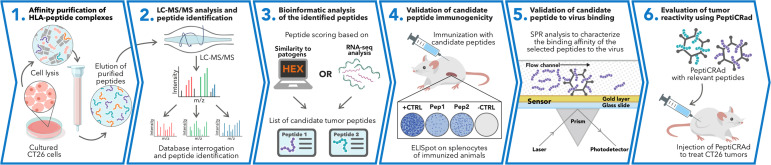
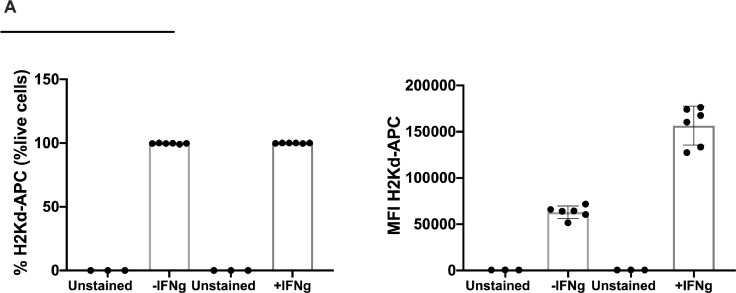
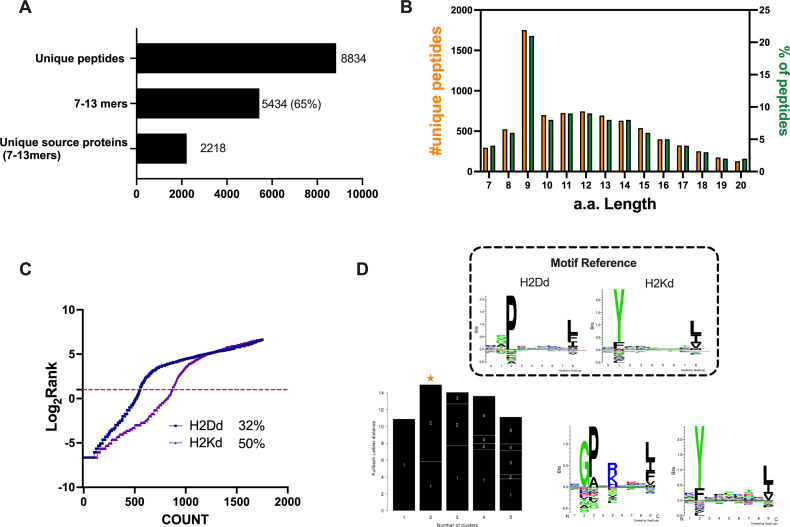
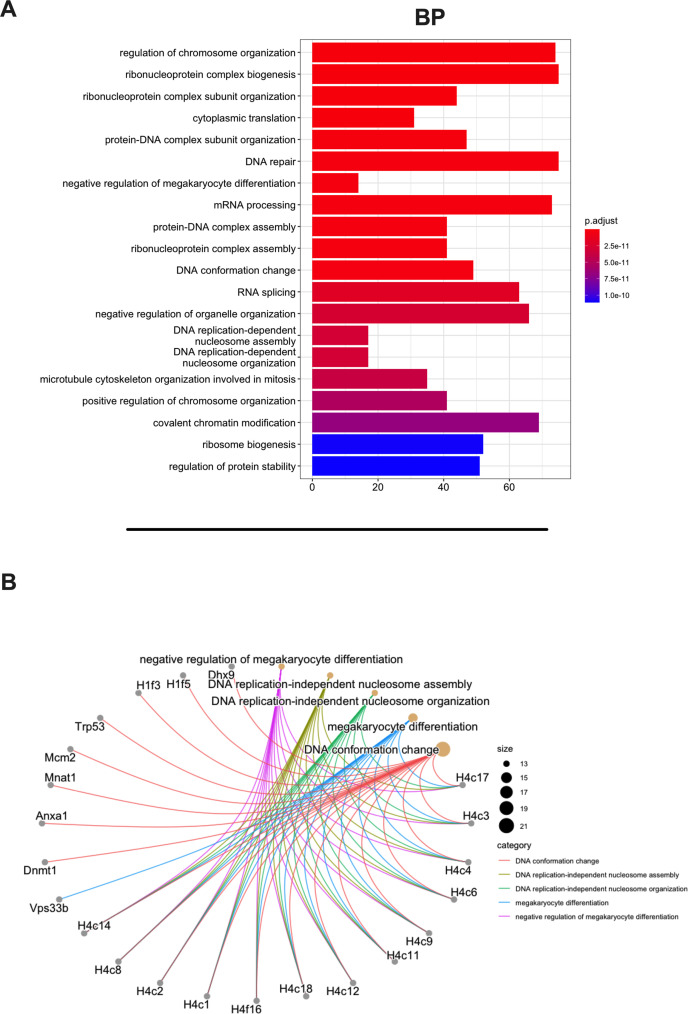
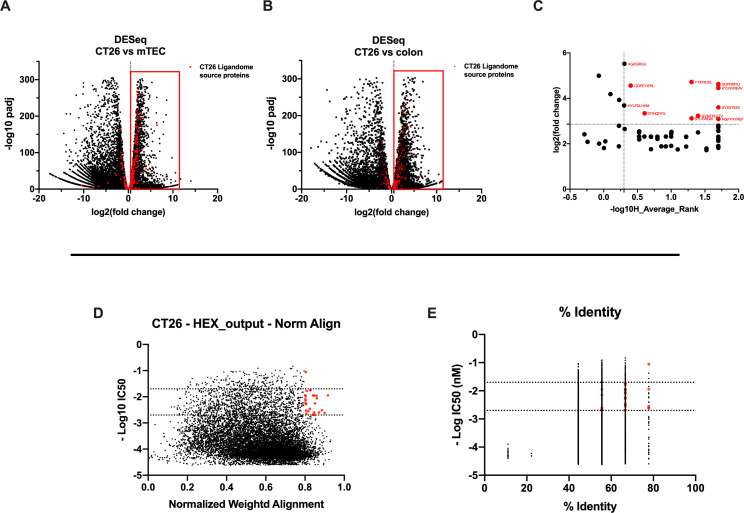
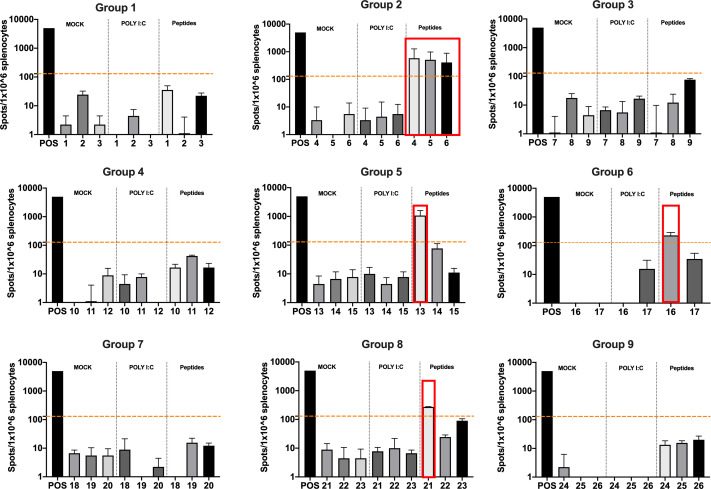
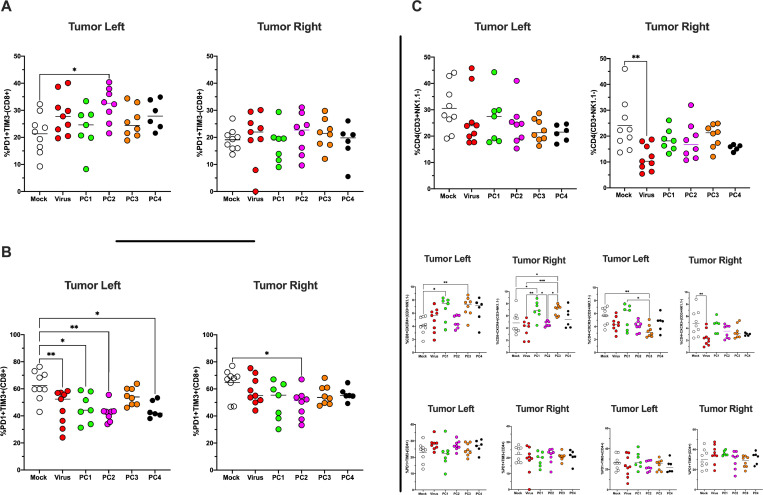
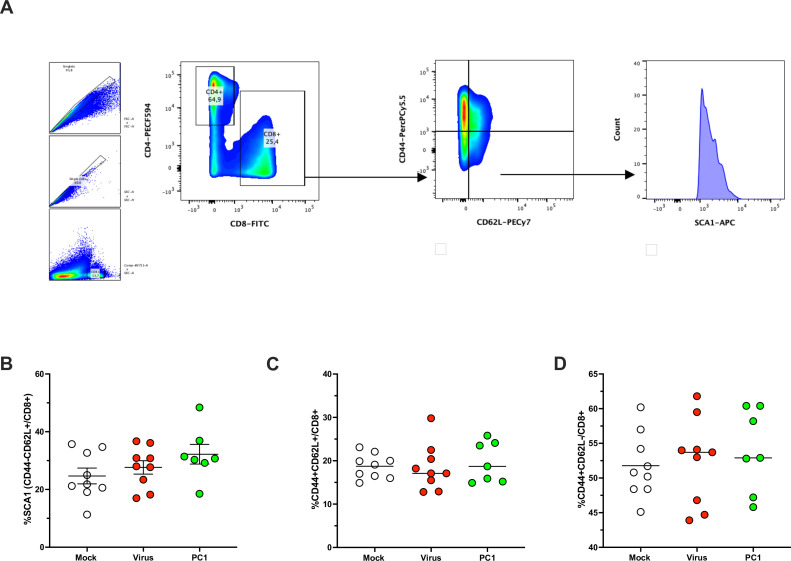
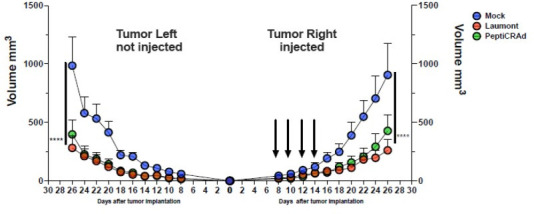
Besides the isolation and identification of major histocompatibility complex I-restricted peptides from the surface of cancer cells, one of the challenges is eliciting an effective antitumor CD8+ T-cell-mediated response as part of therapeutic cancer vaccine. Therefore, the establishment of a solid pipeline for the downstream selection of clinically relevant peptides and the subsequent creation of therapeutic cancer vaccines are of utmost importance. Indeed, the use of peptides for eliciting specific antitumor adaptive immunity is hindered by two main limitations: the efficient selection of the most optimal candidate peptides and the use of a highly immunogenic platform to combine with the peptides to induce effective tumor-specific adaptive immune responses. Here, we describe for the first time a streamlined pipeline for the generation of personalized cancer vaccines starting from the isolation and selection of the most immunogenic peptide candidates expressed on the tumor cells and ending in the generation of efficient therapeutic oncolytic cancer vaccines. This immunopeptidomics-based pipeline was carefully validated in a murine colon tumor model CT26. Specifically, we used state-of-the-art immunoprecipitation and mass spectrometric methodologies to isolate >8000 peptide targets from the CT26 tumor cell line. The selection of the target candidates was then based on two separate approaches: RNAseq analysis and HEX software. The latter is a tool previously developed by Jacopo, 2020, able to identify tumor antigens similar to pathogen antigens in order to exploit molecular mimicry and tumor pathogen cross-reactive T cells in cancer vaccine development. The generated list of candidates (26 in total) was further tested in a functional characterization assay using interferon-γ enzyme-linked immunospot (ELISpot), reducing the number of candidates to six. These peptides were then tested in our previously described oncolytic cancer vaccine platform PeptiCRAd, a vaccine platform that combines an immunogenic oncolytic adenovirus (OAd) coated with tumor antigen peptides. In our work, PeptiCRAd was successfully used for the treatment of mice bearing CT26, controlling the primary malignant lesion and most importantly a secondary, nontreated, cancer lesion. These results confirmed the feasibility of applying the described pipeline for the selection of peptide candidates and generation of therapeutic oncolytic cancer vaccine, filling a gap in the field of cancer immunotherapy, and paving the way to translate our pipeline into human therapeutic approach.
Keywords: immunopetidome; medicine; mouse; onocolytic virus; peptides.
© 2022, Feola et al.
Conflict of interest statement
SF, JC, BM, SR, MF, EY, CB, ER, FH, MF, GA, TV, MG, RB, JL, VC No competing interests declared, SP is an employee and a shareholder at VALO Therapeutics
Figures
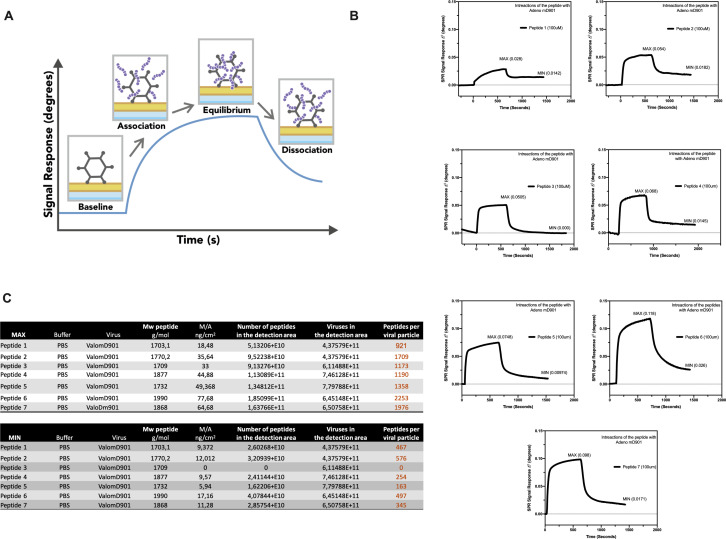
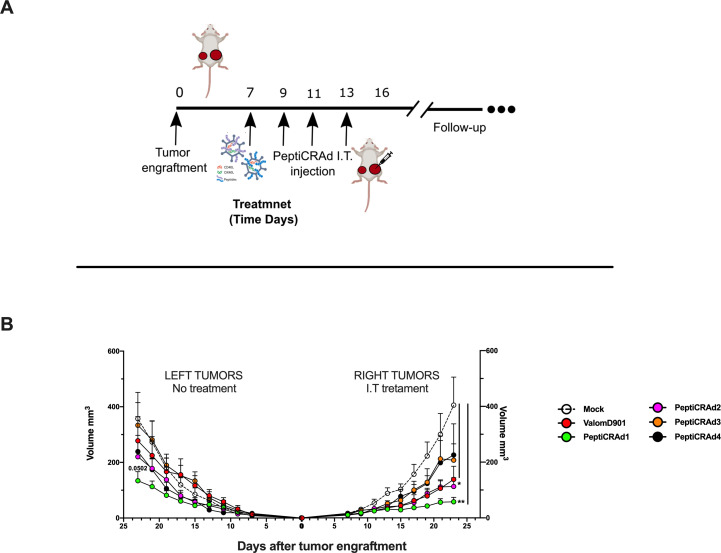
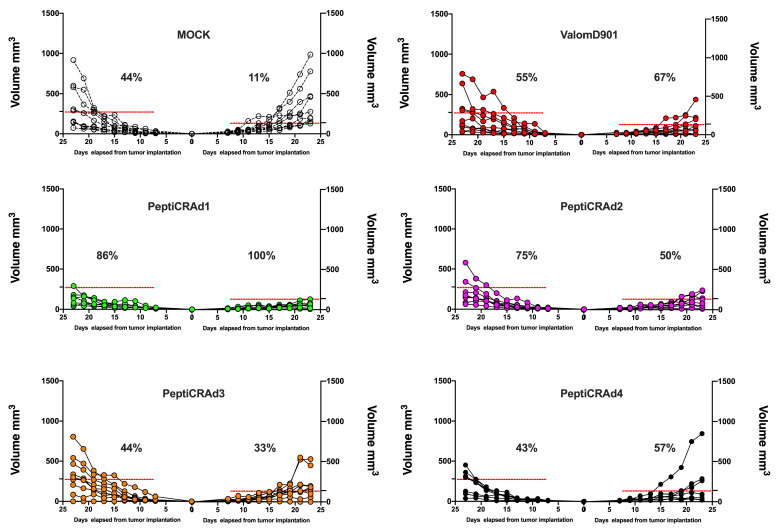
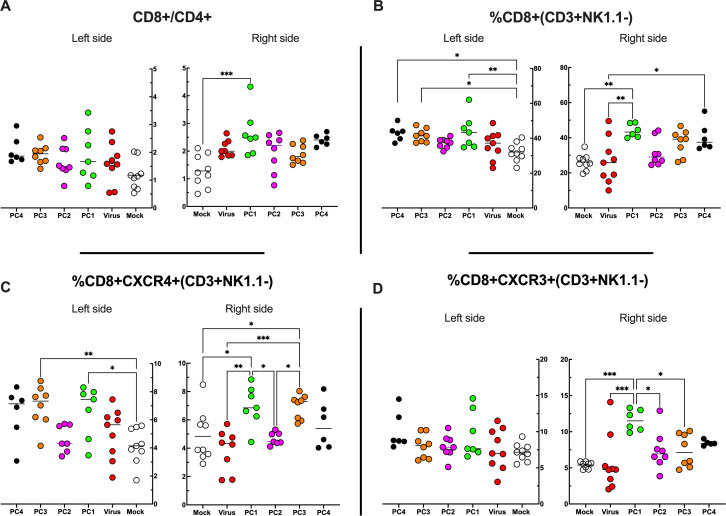
Similar articles
-
Peptides-Coated Oncolytic Vaccines for Cancer Personalized Medicine.Front Immunol. 2022 Apr 14;13:826164. doi: 10.3389/fimmu.2022.826164. eCollection 2022. Front Immunol. 2022. PMID: 35493448 Free PMC article.
-
Exploiting Preexisting Immunity to Enhance Oncolytic Cancer Immunotherapy.Cancer Res. 2020 Jun 15;80(12):2575-2585. doi: 10.1158/0008-5472.CAN-19-2062. Epub 2020 Feb 27. Cancer Res. 2020. PMID: 32107211
-
Virus-like particle-mediated delivery of structure-selected neoantigens demonstrates immunogenicity and antitumoral activity in mice.J Transl Med. 2024 Jan 3;22(1):14. doi: 10.1186/s12967-023-04843-8. J Transl Med. 2024. PMID: 38172991 Free PMC article.
-
Integrating immunopeptidome analysis for the design and development of cancer vaccines.Semin Immunol. 2023 May;67:101750. doi: 10.1016/j.smim.2023.101750. Epub 2023 Mar 30. Semin Immunol. 2023. PMID: 37003057 Review.
-
Sharpening the Edge for Precision Cancer Immunotherapy: Targeting Tumor Antigens through Oncolytic Vaccines.Front Immunol. 2017 Jul 13;8:800. doi: 10.3389/fimmu.2017.00800. eCollection 2017. Front Immunol. 2017. PMID: 28751892 Free PMC article. Review.
Cited by
-
Recent Trends in Nanocarrier-Based Drug Delivery System for Prostate Cancer.AAPS PharmSciTech. 2024 Mar 6;25(3):55. doi: 10.1208/s12249-024-02765-2. AAPS PharmSciTech. 2024. PMID: 38448649 Review.
-
HLAPepBinder: An Ensemble Model for The Prediction Of HLA-Peptide Binding.Iran J Biotechnol. 2024 Oct 1;22(4):e3927. doi: 10.30498/ijb.2024.459448.3927. eCollection 2024 Oct. Iran J Biotechnol. 2024. PMID: 40225296 Free PMC article.
-
Mass Spectrometry-Based Immunopeptidomics of Peptides Presented on Human Leukocyte Antigen Proteins.Methods Mol Biol. 2024;2758:425-443. doi: 10.1007/978-1-0716-3646-6_23. Methods Mol Biol. 2024. PMID: 38549028
-
Cancer vaccines: an update on recent achievements and prospects for cancer therapy.Clin Exp Med. 2024 Dec 25;25(1):24. doi: 10.1007/s10238-024-01541-7. Clin Exp Med. 2024. PMID: 39720956 Free PMC article. Review.
-
Mining the Immunopeptidome for Antigenic Peptides in Cancer.Cancers (Basel). 2022 Oct 11;14(20):4968. doi: 10.3390/cancers14204968. Cancers (Basel). 2022. PMID: 36291752 Free PMC article. Review.
References
-
- Bassani-Sternberg M, Bräunlein E, Klar R, Engleitner T, Sinitcyn P, Audehm S, Straub M, Weber J, Slotta-Huspenina J, Specht K, Martignoni ME, Werner A, Hein R, H Busch D, Peschel C, Rad R, Cox J, Mann M, Krackhardt AM. Direct identification of clinically relevant neoepitopes presented on native human melanoma tissue by mass spectrometry. Nature Communications. 2016;7:13404. doi: 10.1038/ncomms13404. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
