

Microbial communities form rich extracellular metabolomes that foster metabolic interactions and promote drug tolerance
- PMID: 35314781
- PMCID: PMC8975748
- DOI: 10.1038/s41564-022-01072-5
Microbial communities form rich extracellular metabolomes that foster metabolic interactions and promote drug tolerance
Abstract
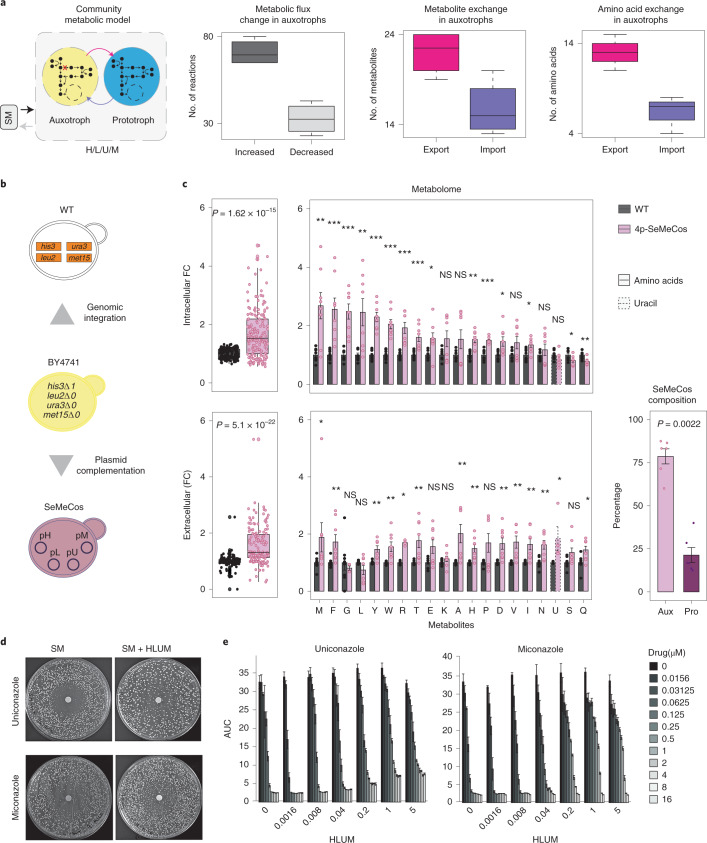
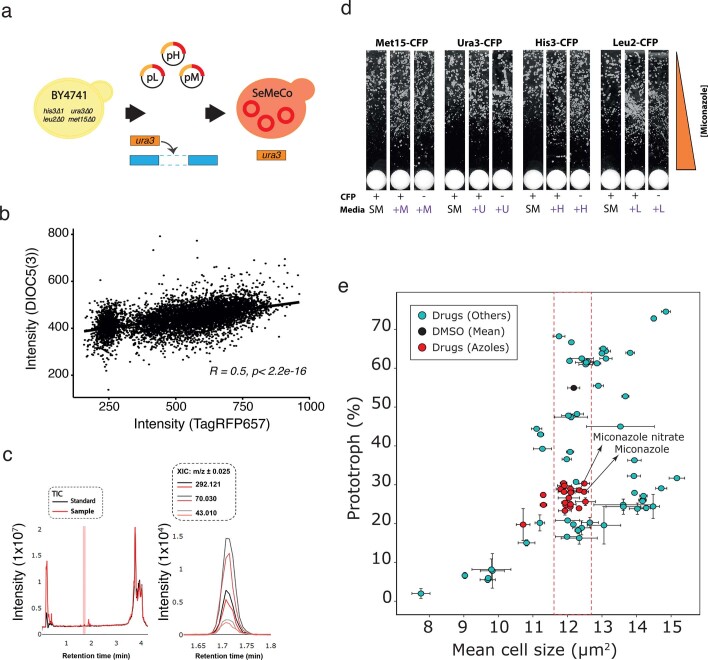
Microbial communities are composed of cells of varying metabolic capacity, and regularly include auxotrophs that lack essential metabolic pathways. Through analysis of auxotrophs for amino acid biosynthesis pathways in microbiome data derived from >12,000 natural microbial communities obtained as part of the Earth Microbiome Project (EMP), and study of auxotrophic-prototrophic interactions in self-establishing metabolically cooperating yeast communities (SeMeCos), we reveal a metabolically imprinted mechanism that links the presence of auxotrophs to an increase in metabolic interactions and gains in antimicrobial drug tolerance. As a consequence of the metabolic adaptations necessary to uptake specific metabolites, auxotrophs obtain altered metabolic flux distributions, export more metabolites and, in this way, enrich community environments in metabolites. Moreover, increased efflux activities reduce intracellular drug concentrations, allowing cells to grow in the presence of drug levels above minimal inhibitory concentrations. For example, we show that the antifungal action of azoles is greatly diminished in yeast cells that uptake metabolites from a metabolically enriched environment. Our results hence provide a mechanism that explains why cells are more robust to drug exposure when they interact metabolically.
© 2022. The Author(s).
Conflict of interest statement
K.C. is employed by AstraZeneca. The other authors declare no competing interests.
Figures












Comment in
-
Give and take in the exometabolome.Nat Microbiol. 2022 Apr;7(4):484-485. doi: 10.1038/s41564-022-01081-4. Nat Microbiol. 2022. PMID: 35314782 No abstract available.
-
Metabolic interactions shape a community's phenotype.Trends Microbiol. 2022 Jul;30(7):609-611. doi: 10.1016/j.tim.2022.05.001. Epub 2022 May 24. Trends Microbiol. 2022. PMID: 35618541
References
-
- Pinu FR, et al. Metabolite secretion in microorganisms: the theory of metabolic overflow put to the test. Metabolomics. 2018;14:43. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
