

KRAS is vulnerable to reversible switch-II pocket engagement in cells
- PMID: 35314814
- PMCID: PMC9135634
- DOI: 10.1038/s41589-022-00985-w
KRAS is vulnerable to reversible switch-II pocket engagement in cells
Abstract
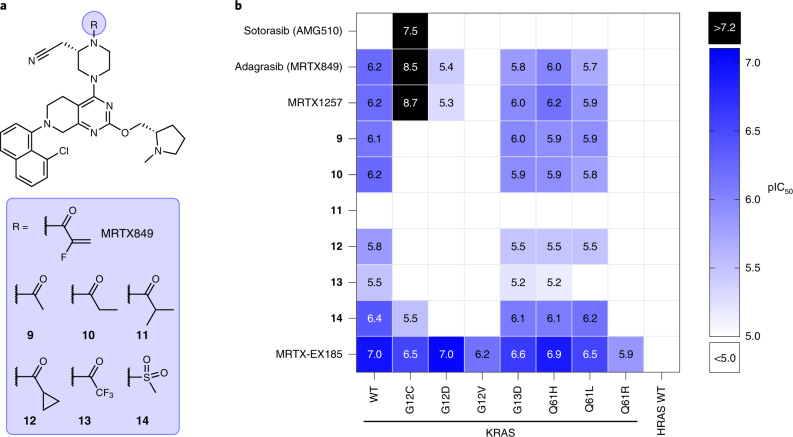
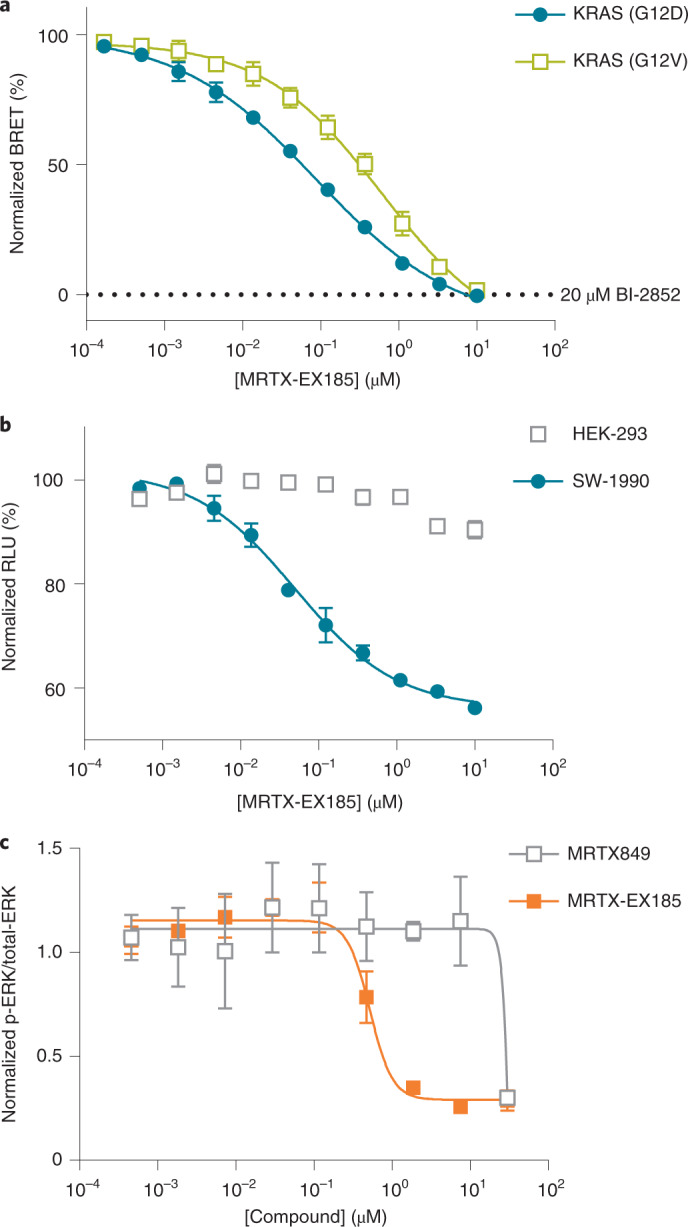
Current small-molecule inhibitors of KRAS(G12C) bind irreversibly in the switch-II pocket (SII-P), exploiting the strong nucleophilicity of the acquired cysteine as well as the preponderance of the GDP-bound form of this mutant. Nevertheless, many oncogenic KRAS mutants lack these two features, and it remains unknown whether targeting the SII-P is a practical therapeutic approach for KRAS mutants beyond G12C. Here we use NMR spectroscopy and a cellular KRAS engagement assay to address this question by examining a collection of SII-P ligands from the literature and from our own laboratory. We show that the SII-Ps of many KRAS hotspot (G12, G13, Q61) mutants are accessible using noncovalent ligands, and that this accessibility is not necessarily coupled to the GDP state of KRAS. The results we describe here emphasize the SII-P as a privileged drug-binding site on KRAS and unveil new therapeutic opportunities in RAS-driven cancer.
© 2022. The Author(s).
Conflict of interest statement
J.D.V., J.A.W., C.A.Z., M.R.T., M.T.B., B.F.B., C.R.C. and M.B.R. are employees of Promega Corporation, which holds patents related to the NanoBRET Target Engagement method. K.M.S. is an inventor on patents owned by University of California San Francisco covering KRAS targeting small molecules licensed to Araxes and Erasca. K.M.S. has consulting agreements for the following companies, which involve monetary and/or stock compensation: Revolution Medicines, Black Diamond Therapeutics, BridGene Biosciences, Denali Therapeutics, Dice Molecules, eFFECTOR Therapeutics, Erasca, Genentech/Roche, Janssen Pharmaceuticals, Kumquat Biosciences, Kura Oncology, Mitokinin, Type6 Therapeutics, Venthera, Wellspring Biosciences (Araxes Pharma), Turning Point, Ikena, Initial Therapeutics and BioTheryX.
Figures
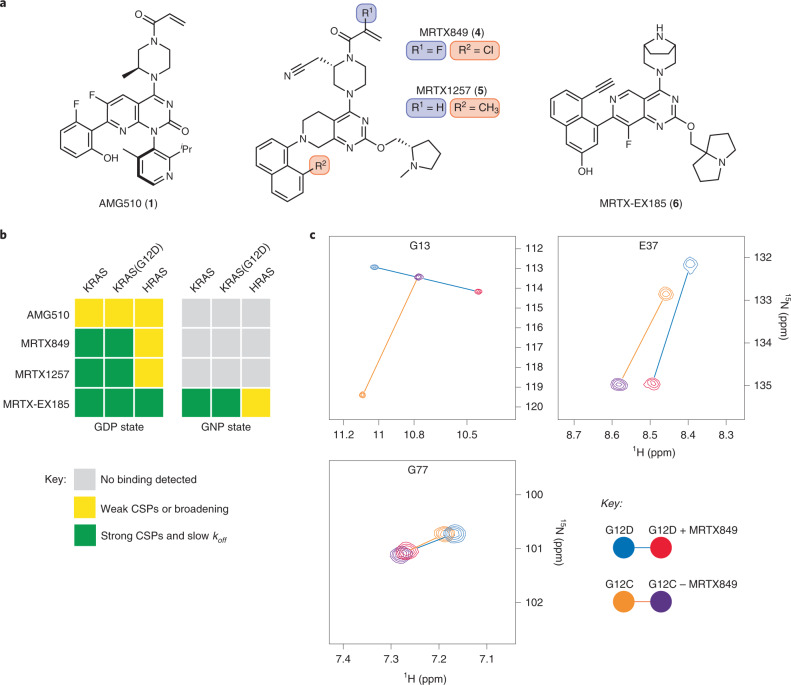
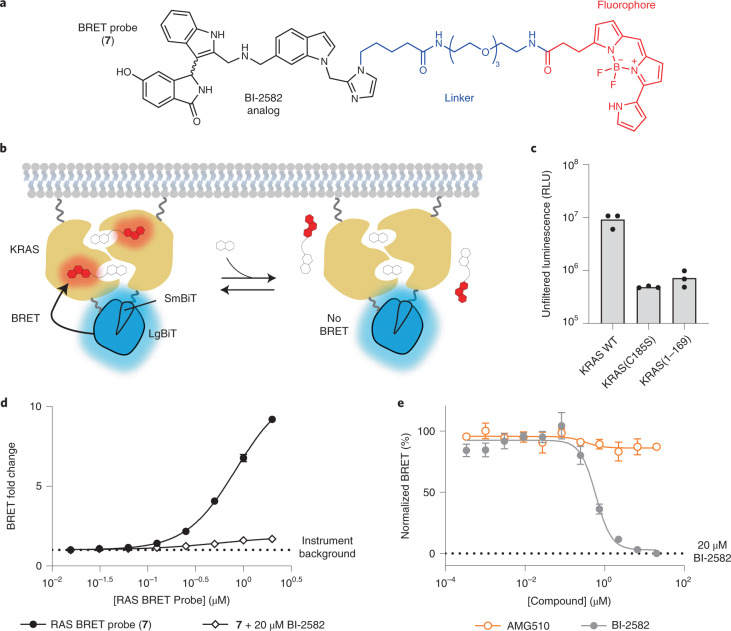
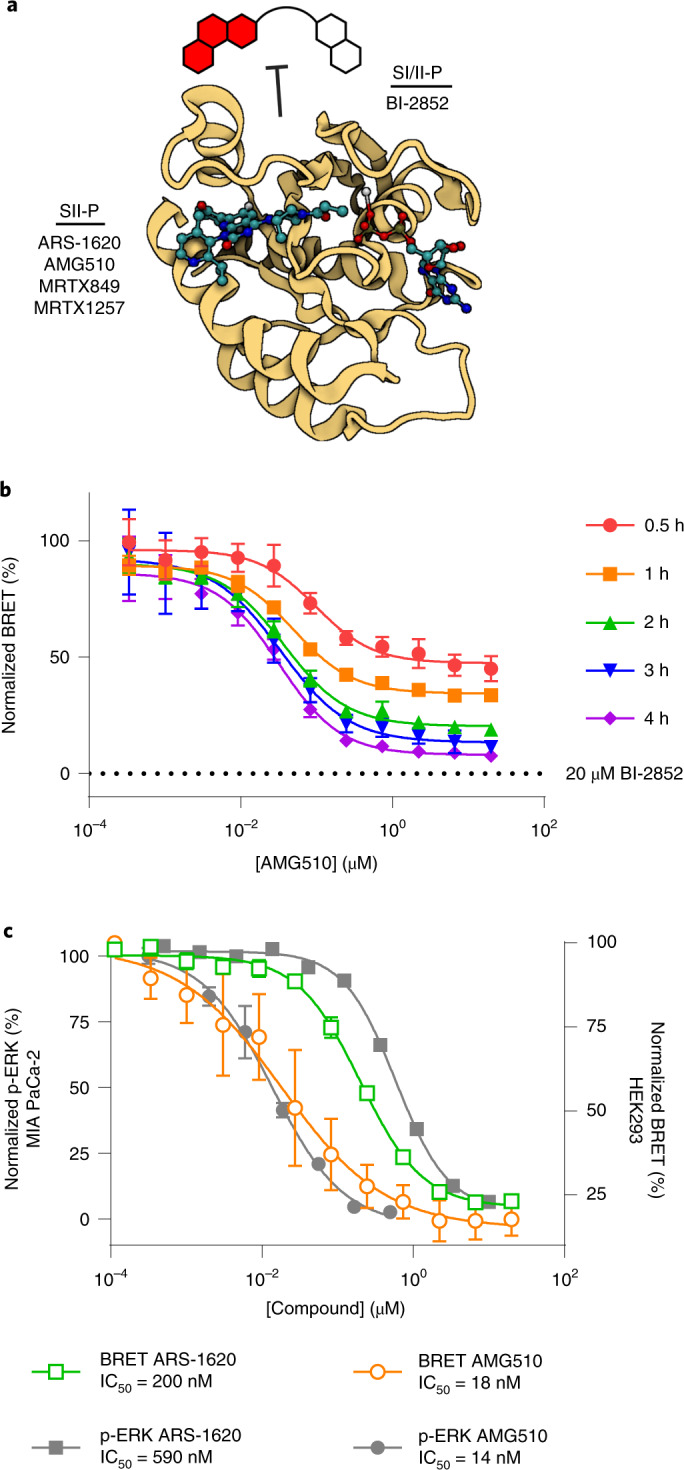
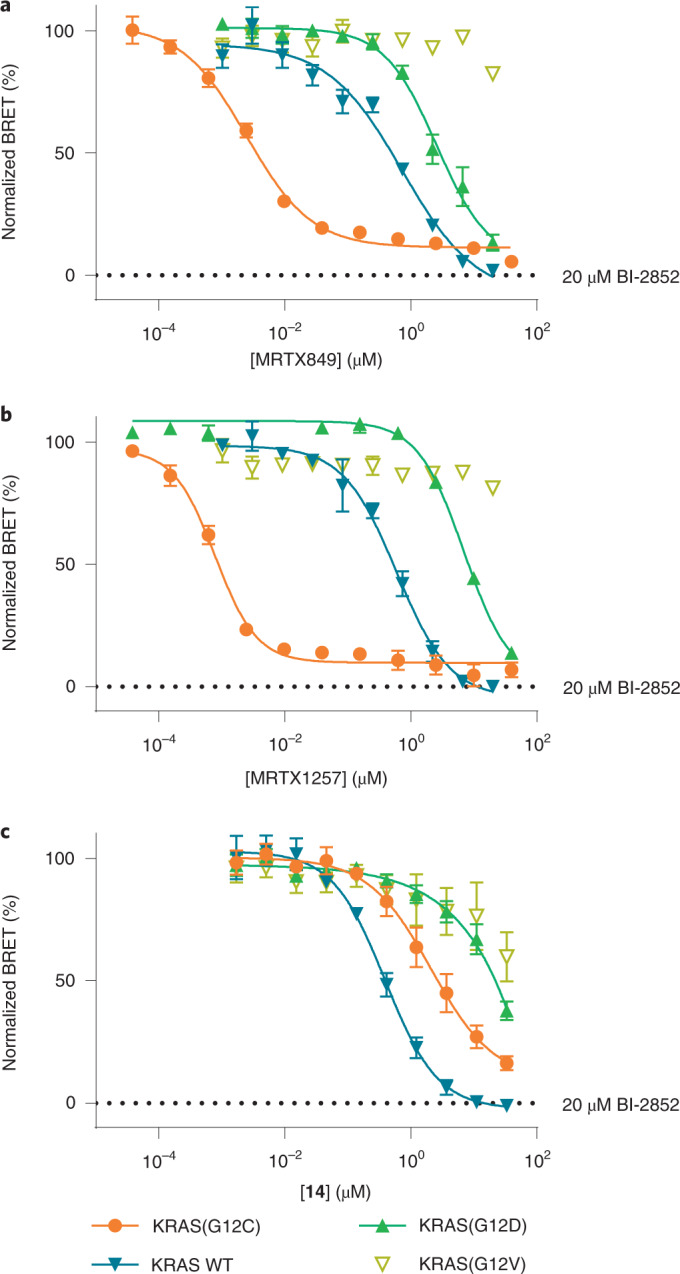
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
