

Spatial transcriptomic reconstruction of the mouse olfactory glomerular map suggests principles of odor processing
- PMID: 35314823
- PMCID: PMC9281876
- DOI: 10.1038/s41593-022-01030-8
Spatial transcriptomic reconstruction of the mouse olfactory glomerular map suggests principles of odor processing
Abstract
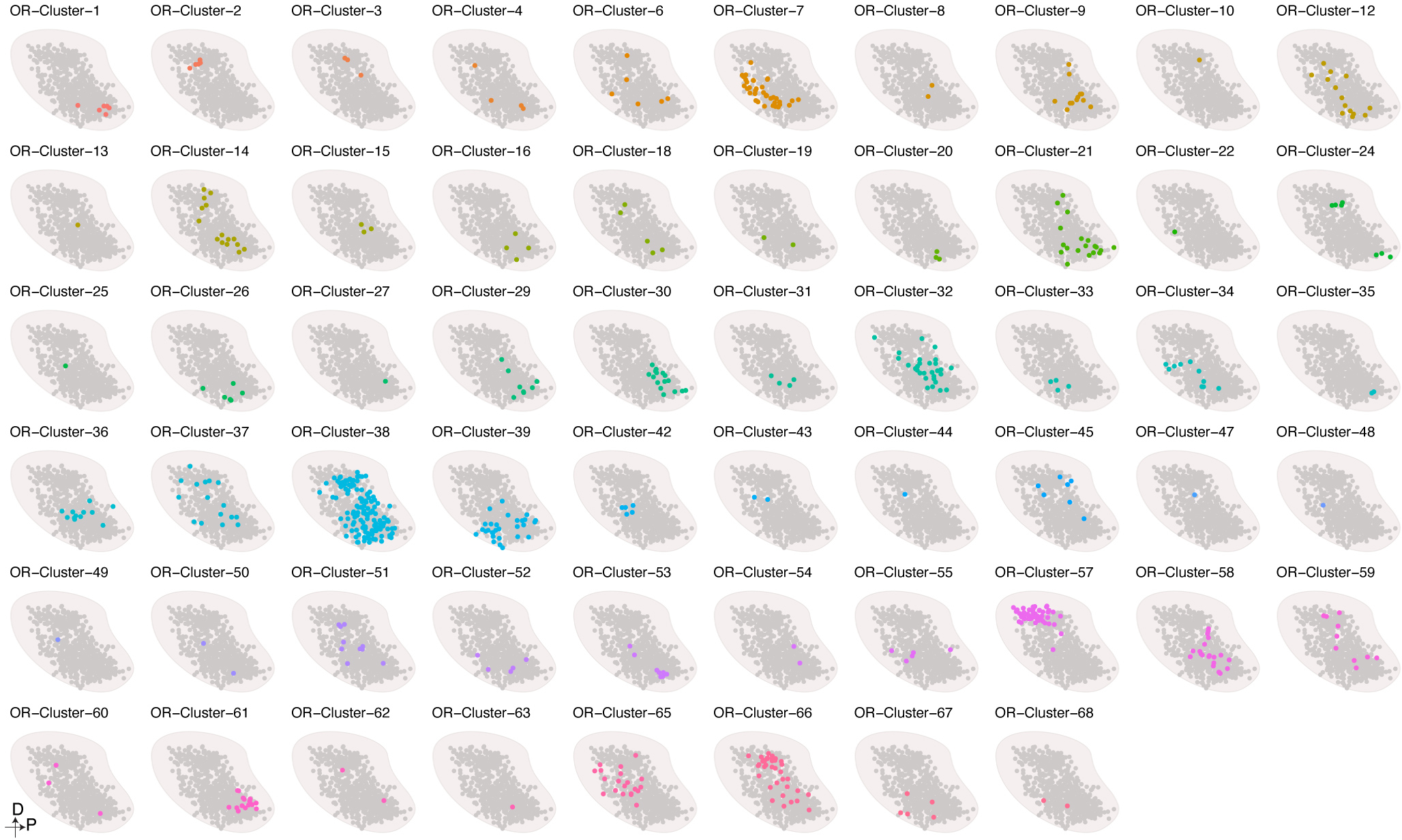
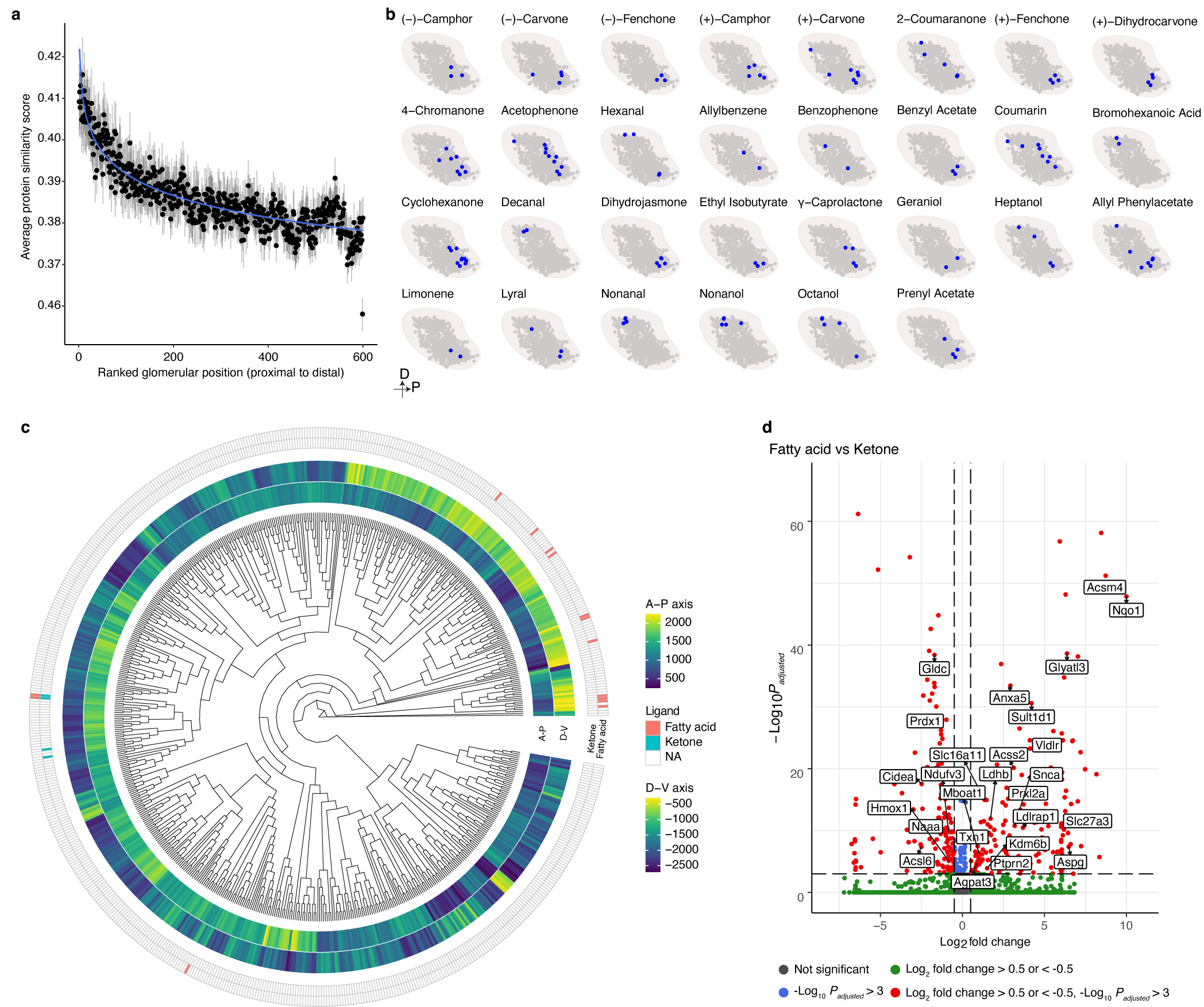
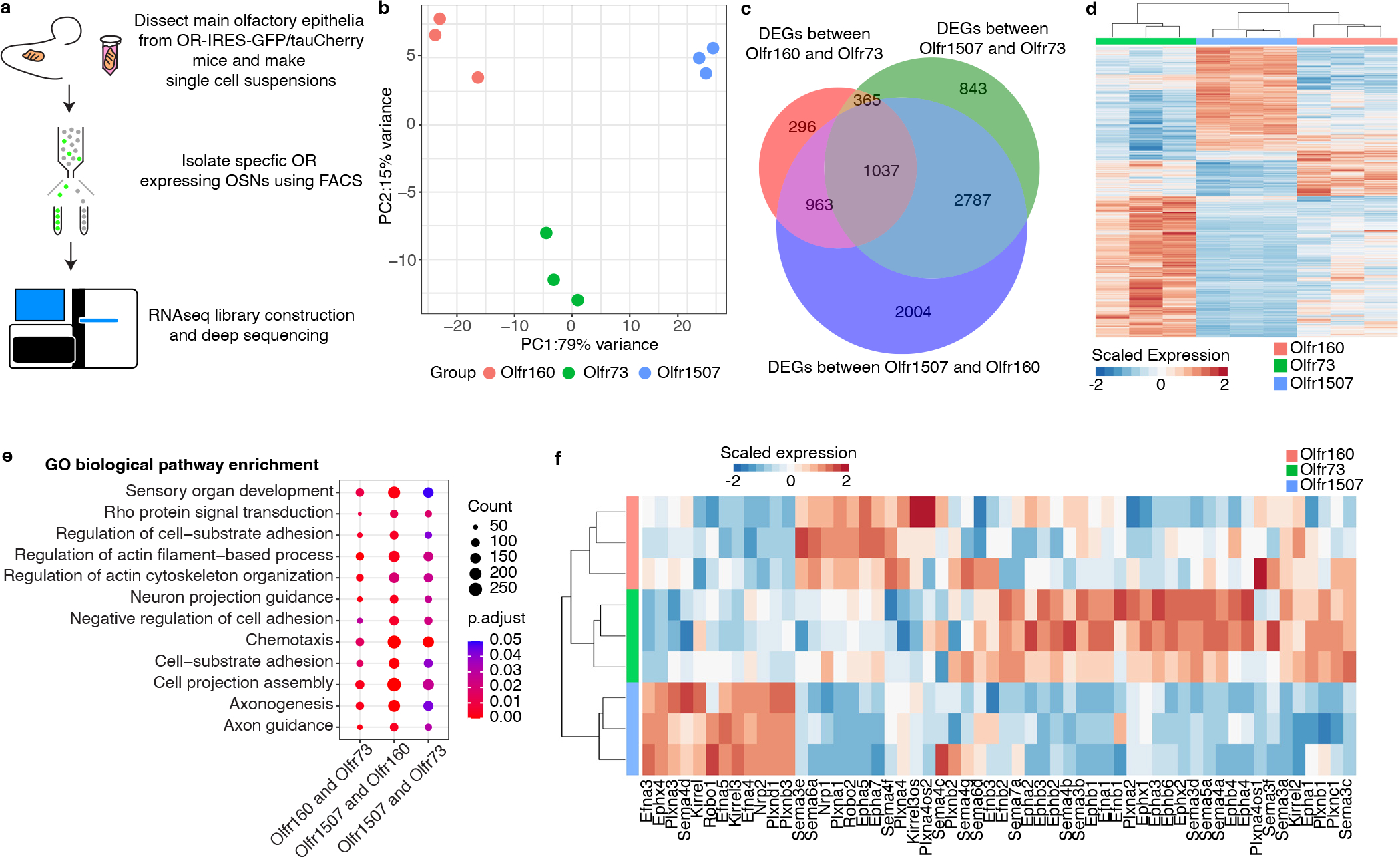
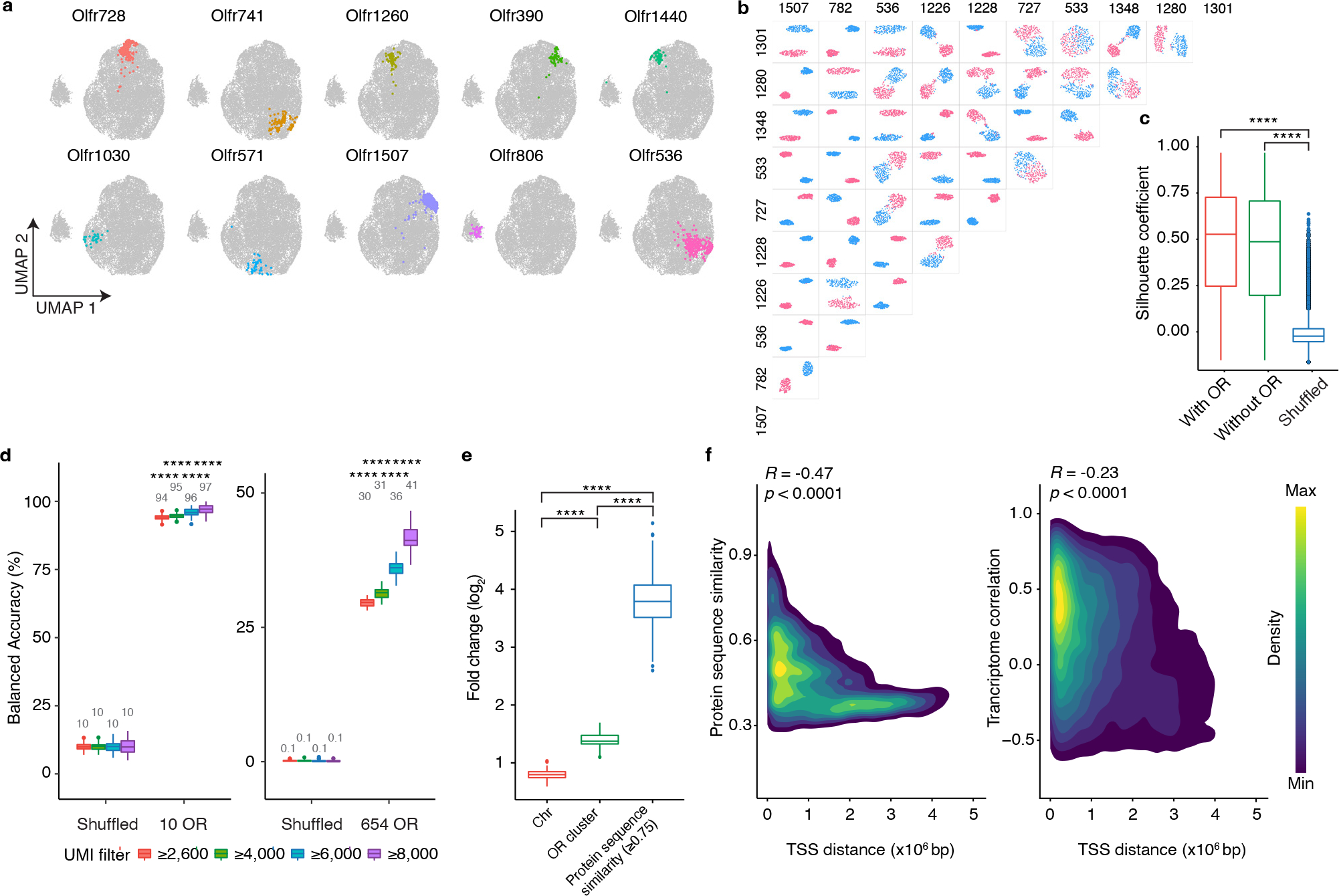
The olfactory system's ability to detect and discriminate between the vast array of chemicals present in the environment is critical for an animal's survival. In mammals, the first step of this odor processing is executed by olfactory sensory neurons, which project their axons to a stereotyped location in the olfactory bulb (OB) to form glomeruli. The stereotyped positioning of glomeruli in the OB suggests an importance for this organization in odor perception. However, because the location of only a limited subset of glomeruli has been determined, it has been challenging to determine the relationship between glomerular location and odor discrimination. Using a combination of single-cell RNA sequencing, spatial transcriptomics and machine learning, we have generated a map of most glomerular positions in the mouse OB. These observations significantly extend earlier studies and suggest an overall organizational principle in the OB that may be used by the brain to assist in odor decoding.
© 2022. The Author(s), under exclusive licence to Springer Nature America, Inc.
Conflict of interest statement
Competing interests
The authors report no competing interests.
Figures
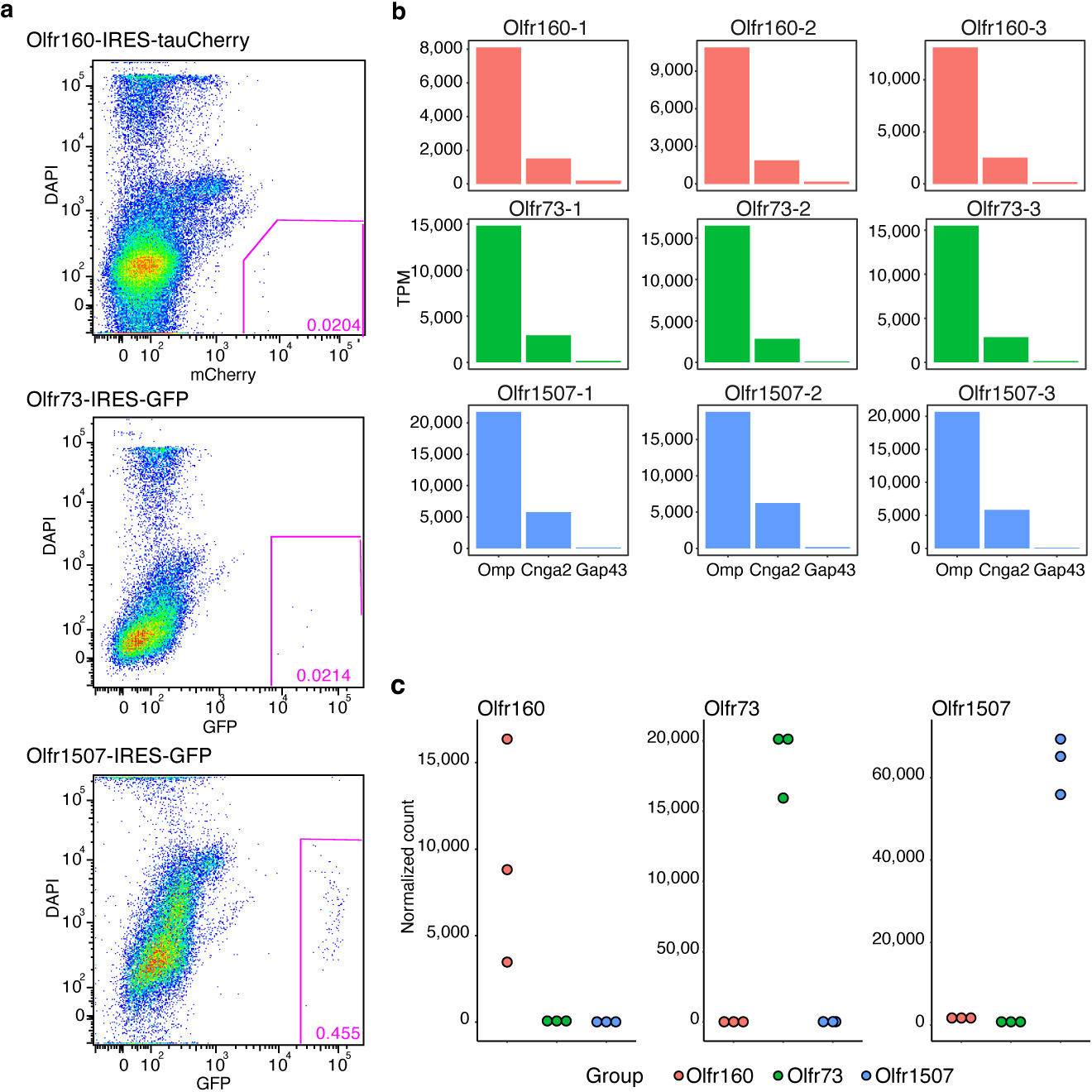
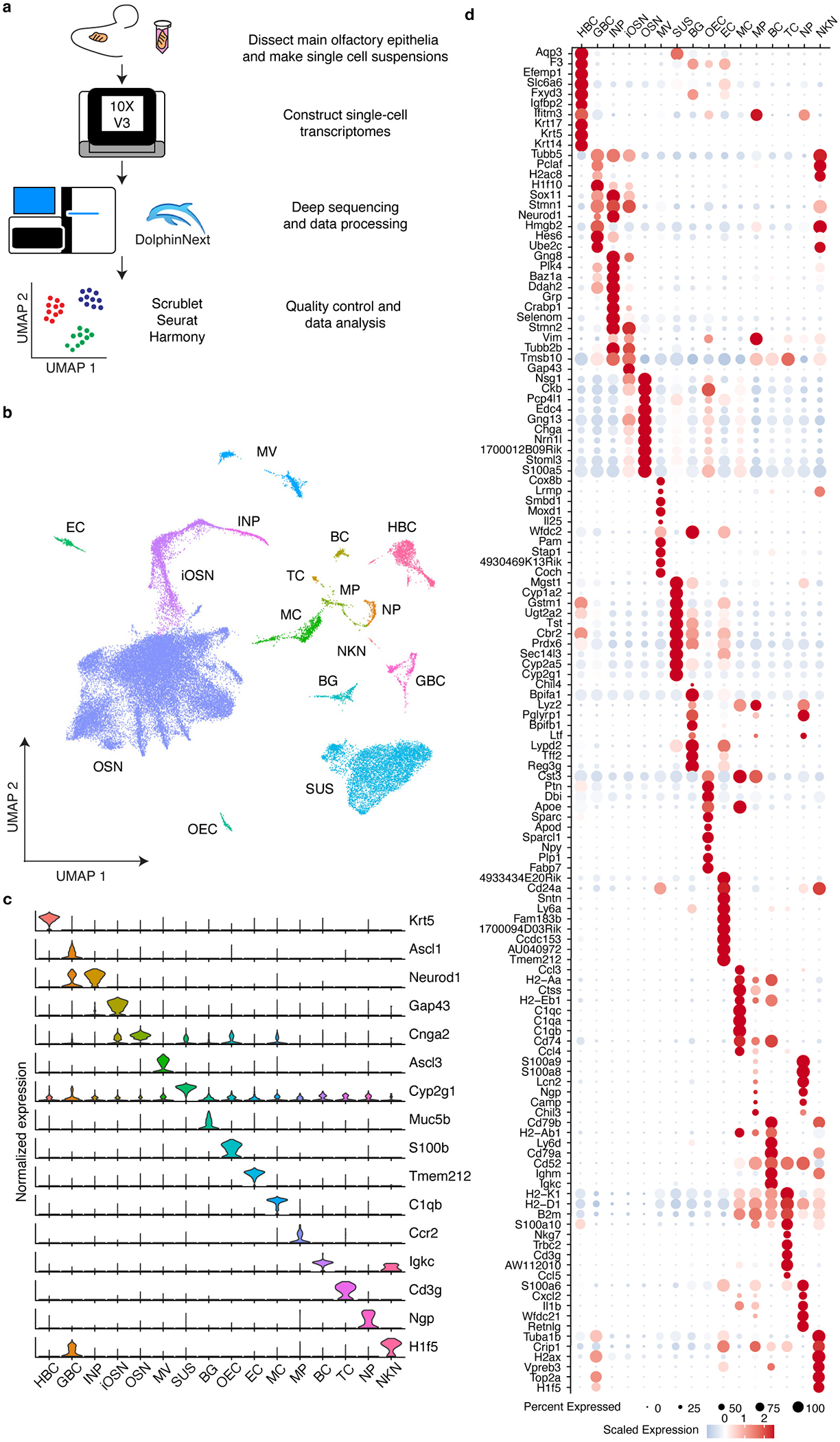
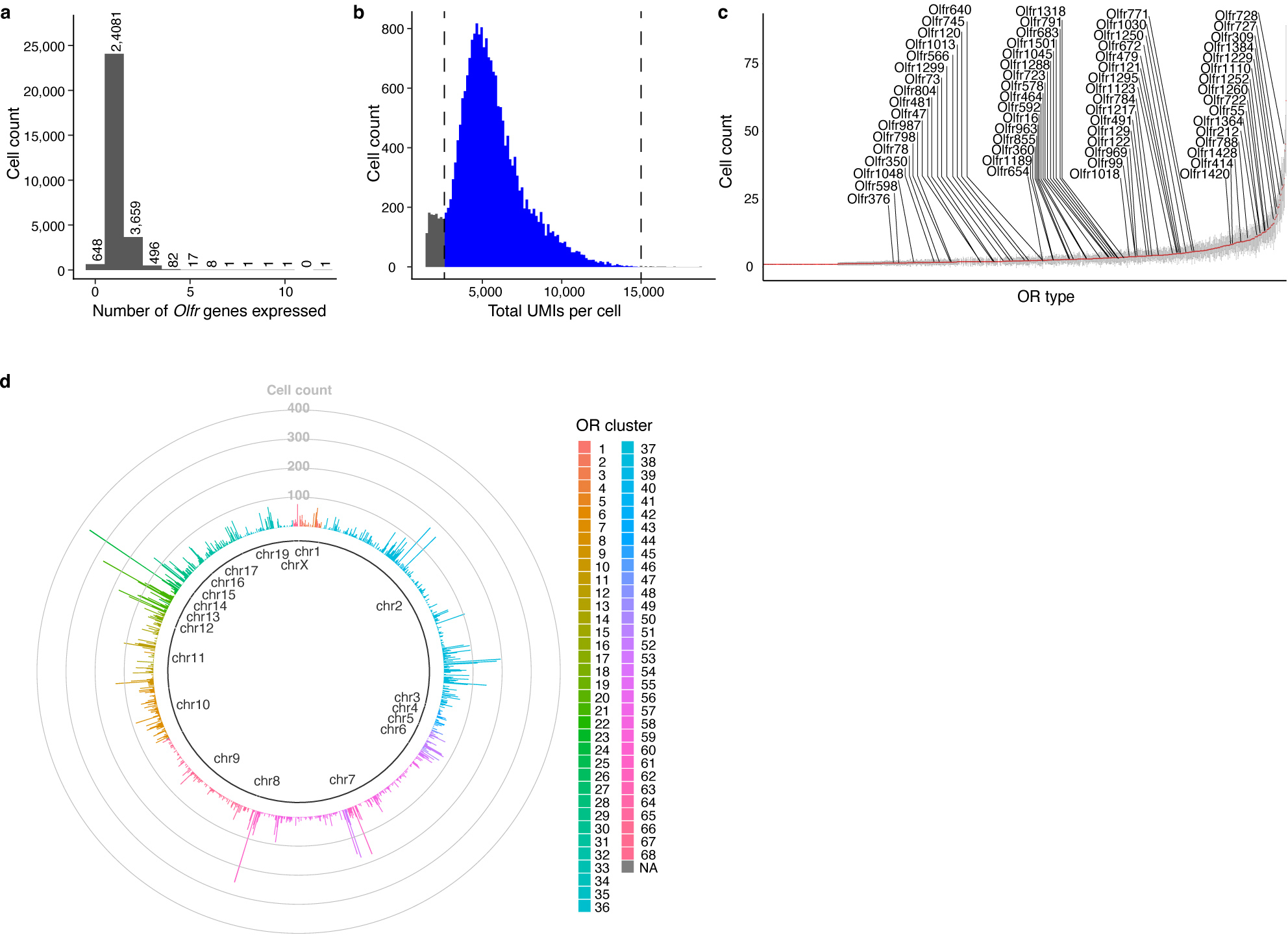
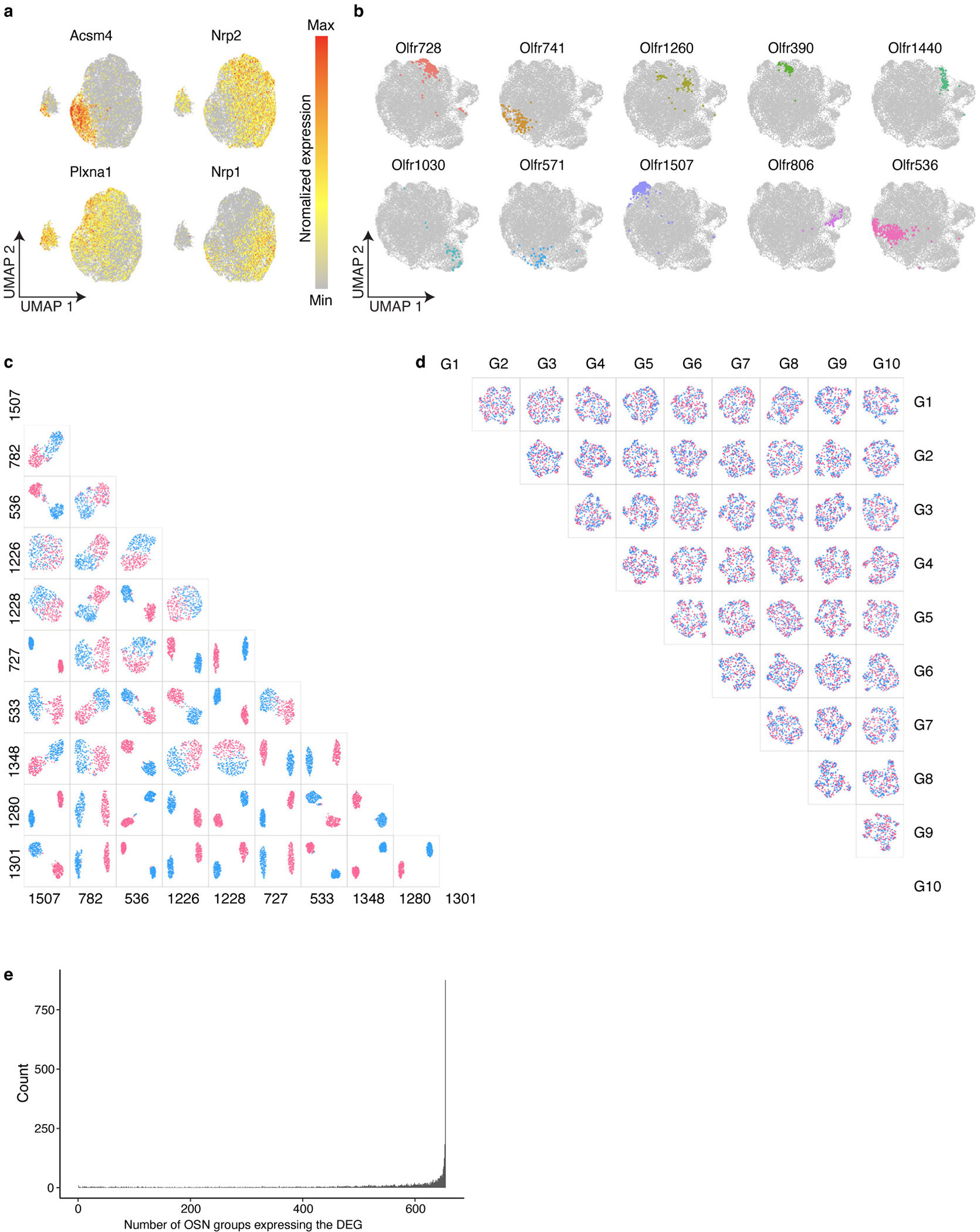
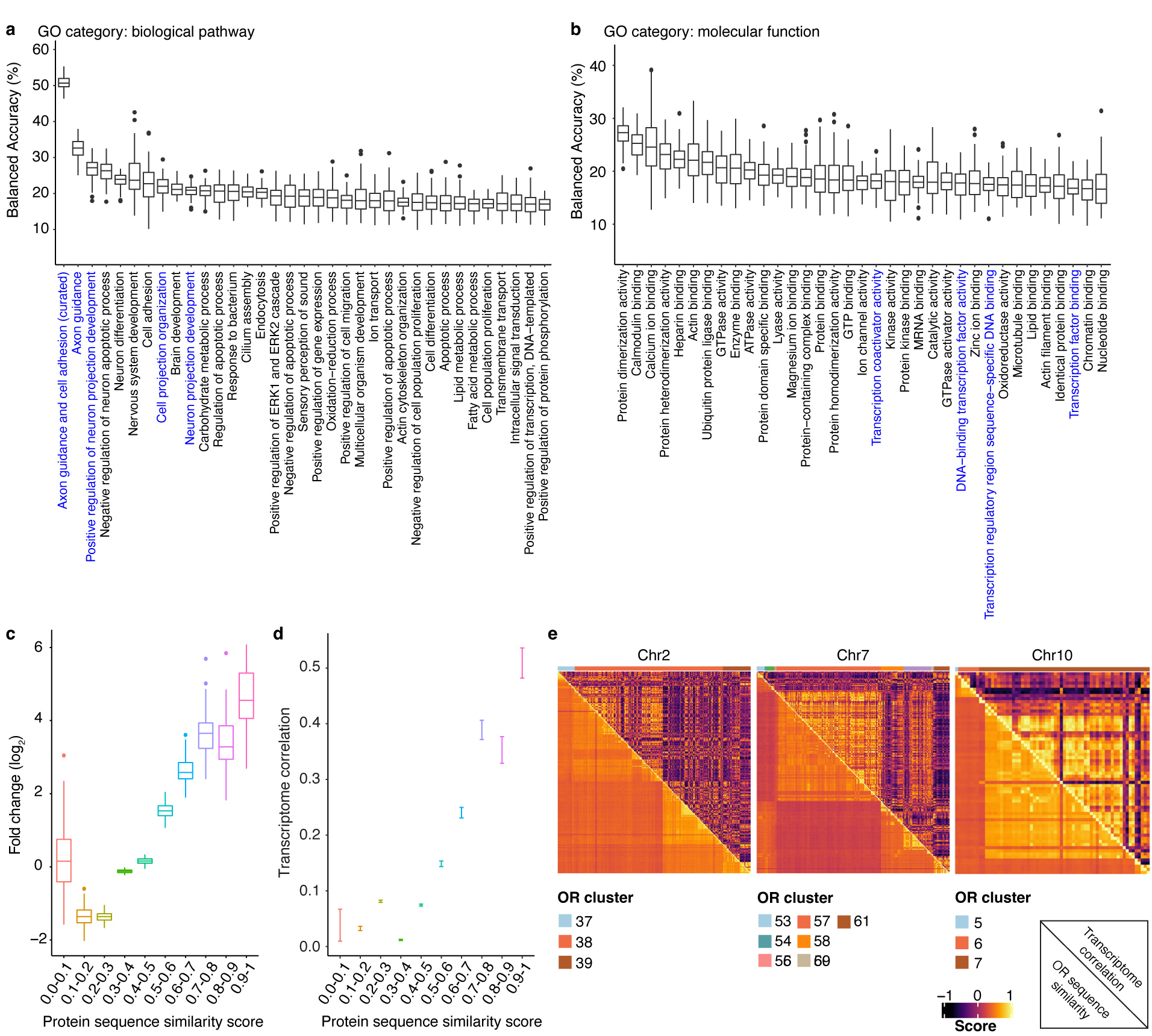
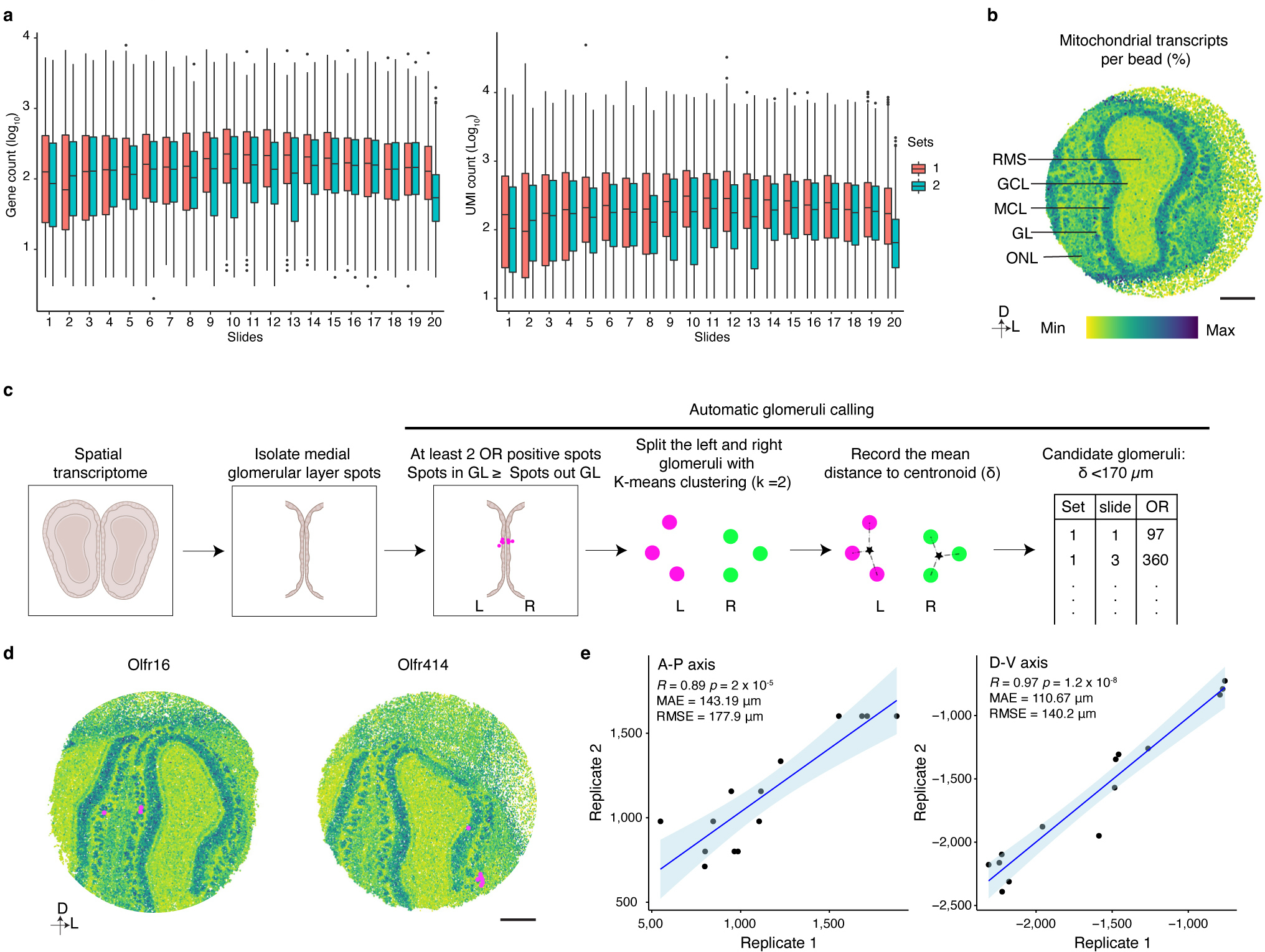
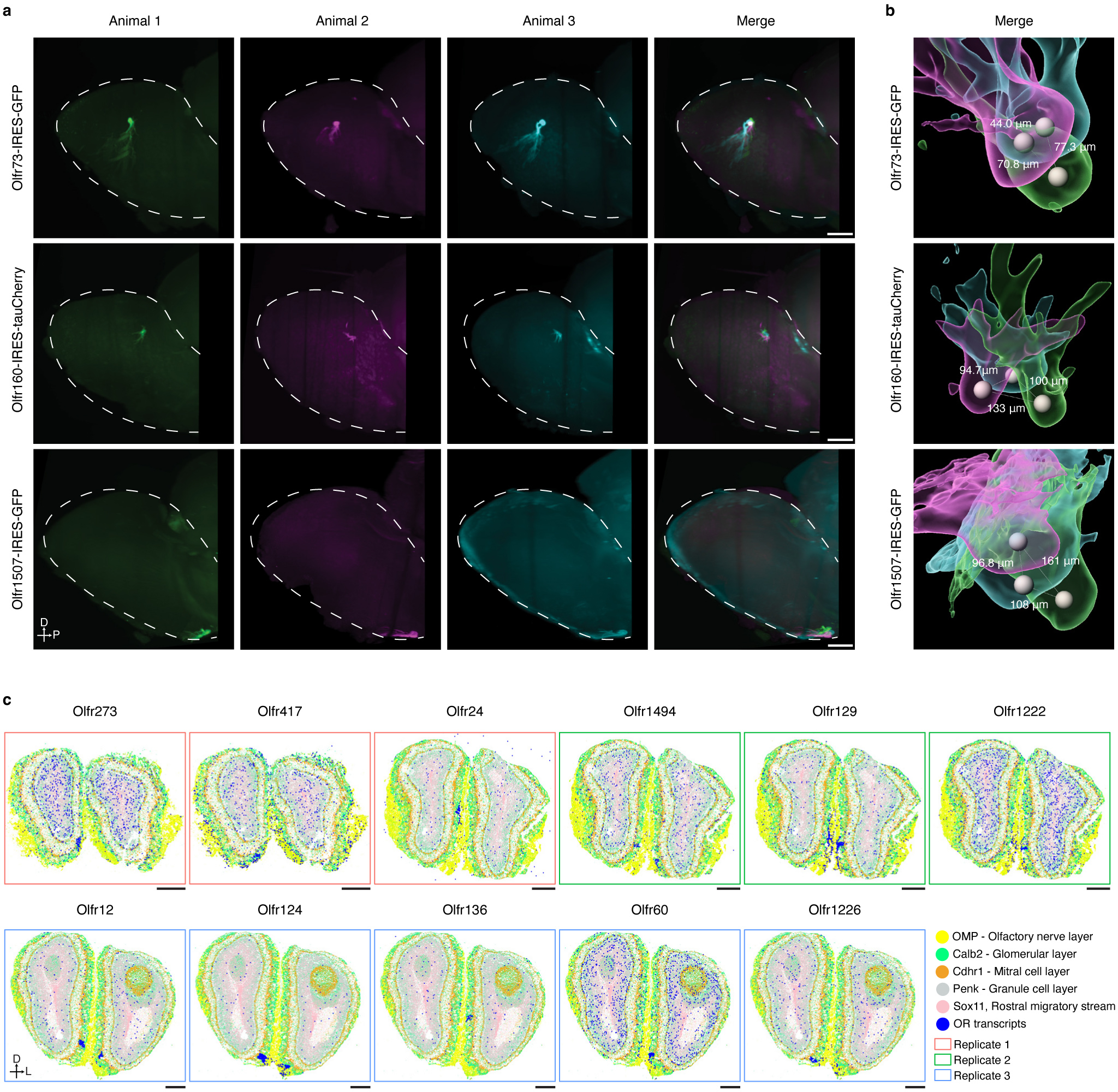
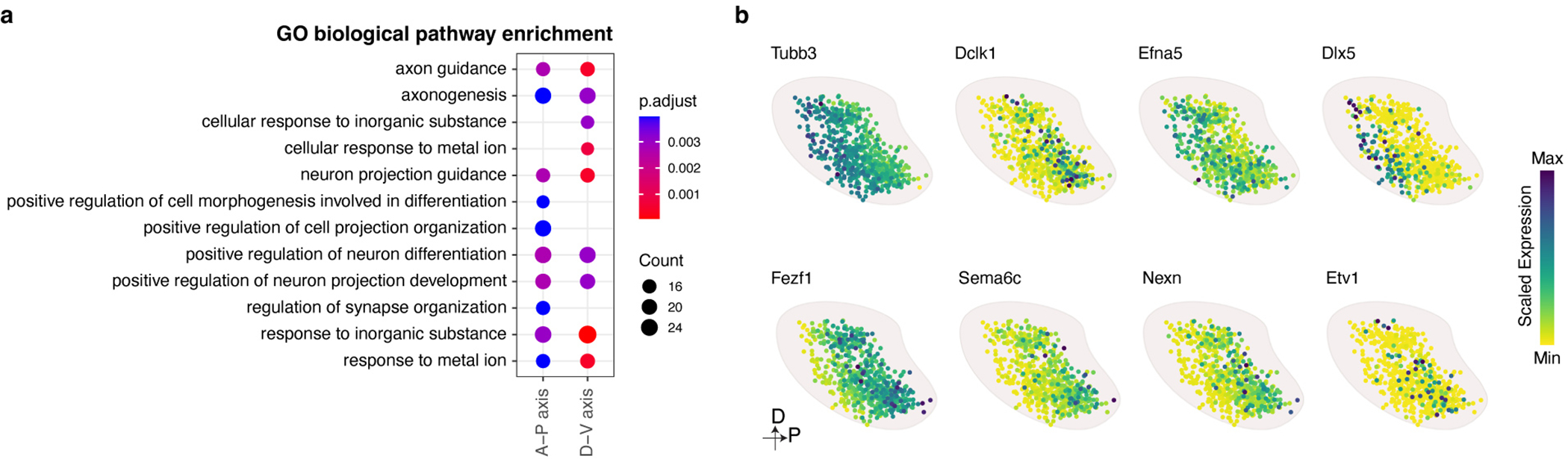
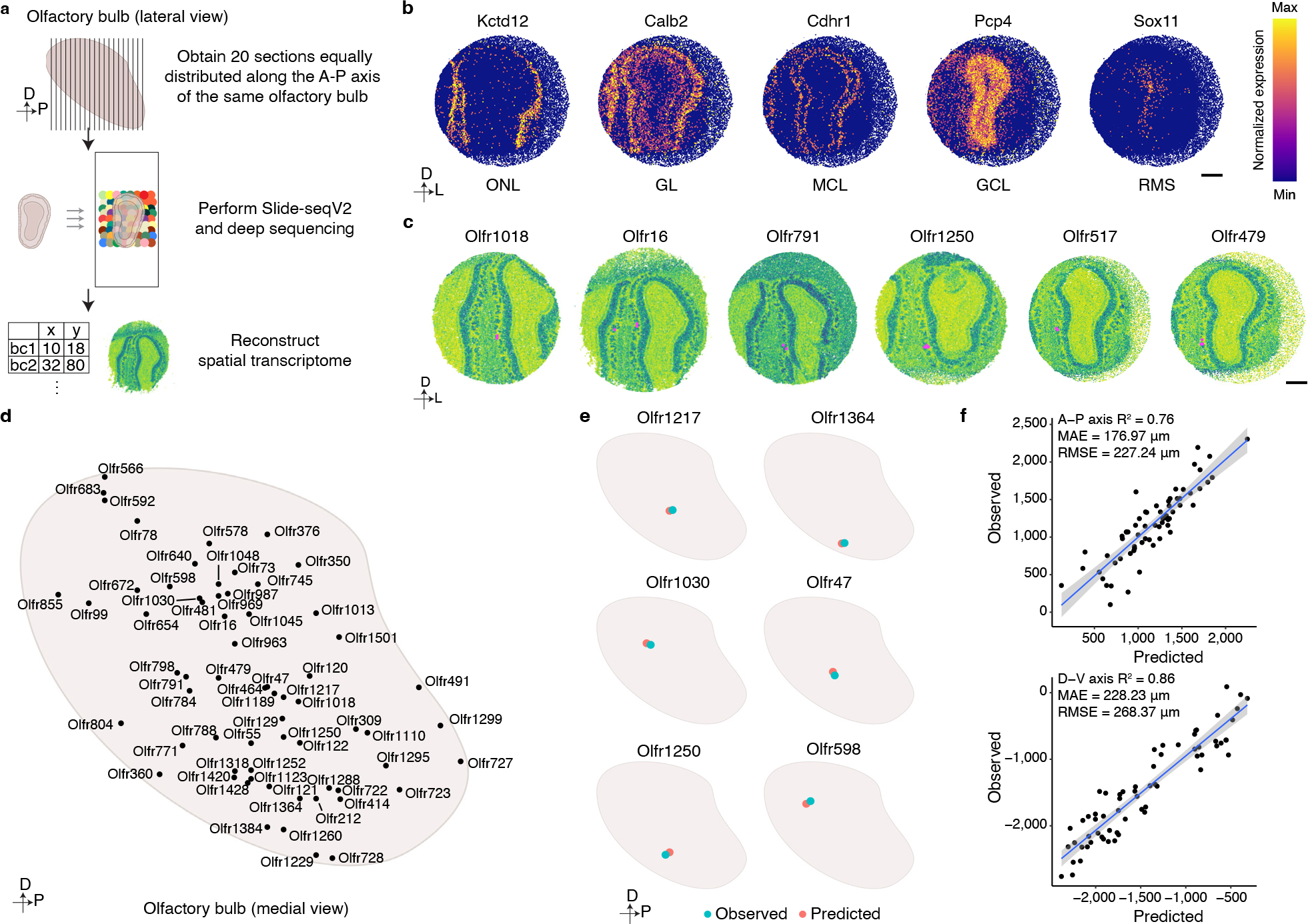
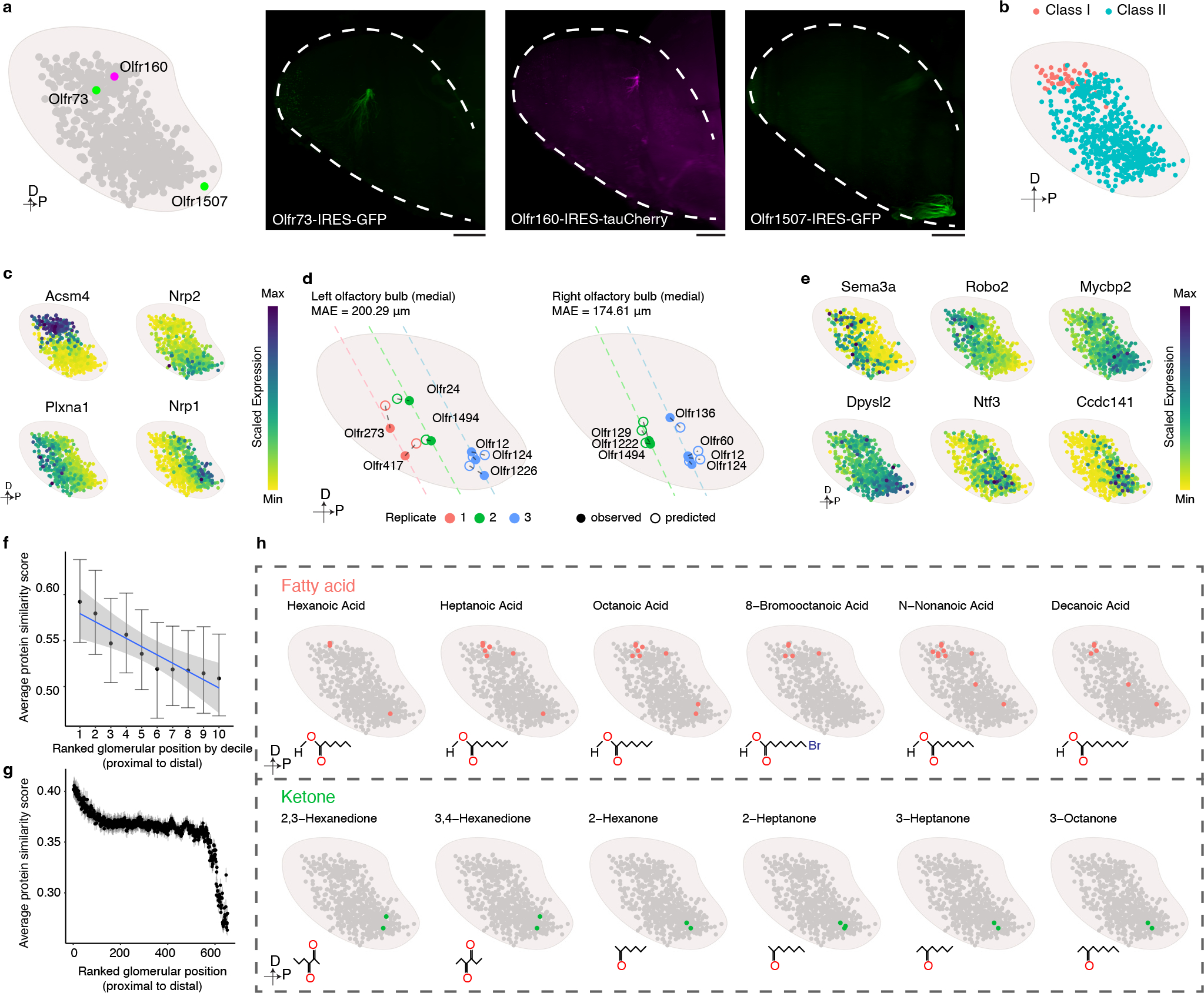
Comment in
-
Mapping odorant receptors to their glomeruli.Nat Neurosci. 2022 Apr;25(4):405-407. doi: 10.1038/s41593-022-01045-1. Nat Neurosci. 2022. PMID: 35314824 No abstract available.
Similar articles
-
Distorted odor maps in the olfactory bulb of semaphorin 3A-deficient mice.J Neurosci. 2003 Feb 15;23(4):1390-7. doi: 10.1523/JNEUROSCI.23-04-01390.2003. J Neurosci. 2003. PMID: 12598627 Free PMC article.
-
Spatiotemporal dynamics of odor responses in the lateral and dorsal olfactory bulb.PLoS Biol. 2019 Sep 18;17(9):e3000409. doi: 10.1371/journal.pbio.3000409. eCollection 2019 Sep. PLoS Biol. 2019. PMID: 31532763 Free PMC article.
-
Principles of glomerular organization in the human olfactory bulb--implications for odor processing.PLoS One. 2008 Jul 9;3(7):e2640. doi: 10.1371/journal.pone.0002640. PLoS One. 2008. PMID: 18612420 Free PMC article.
-
The olfactory bulb: coding and processing of odor molecule information.Science. 1999 Oct 22;286(5440):711-5. doi: 10.1126/science.286.5440.711. Science. 1999. PMID: 10531048 Review.
-
Odor representations in the mammalian olfactory bulb.Wiley Interdiscip Rev Syst Biol Med. 2010 Sep-Oct;2(5):603-611. doi: 10.1002/wsbm.85. Wiley Interdiscip Rev Syst Biol Med. 2010. PMID: 20836051 Review.
Cited by
-
ER stress transforms random olfactory receptor choice into axon targeting precision.Cell. 2022 Oct 13;185(21):3896-3912.e22. doi: 10.1016/j.cell.2022.08.025. Epub 2022 Sep 26. Cell. 2022. PMID: 36167070 Free PMC article.
-
GPRC5C regulates the composition of cilia in the olfactory system.BMC Biol. 2023 Dec 18;21(1):292. doi: 10.1186/s12915-023-01790-0. BMC Biol. 2023. PMID: 38110903 Free PMC article.
-
Challenges and Opportunities for Immunoprofiling Using a Spatial High-Plex Technology: The NanoString GeoMx® Digital Spatial Profiler.Front Oncol. 2022 Jun 29;12:890410. doi: 10.3389/fonc.2022.890410. eCollection 2022. Front Oncol. 2022. PMID: 35847846 Free PMC article. Review.
-
spatiAlign: an unsupervised contrastive learning model for data integration of spatially resolved transcriptomics.Gigascience. 2024 Jan 2;13:giae042. doi: 10.1093/gigascience/giae042. Gigascience. 2024. PMID: 39028588 Free PMC article.
-
Benchmarking algorithms for spatially variable gene identification in spatial transcriptomics.Bioinformatics. 2025 Mar 29;41(4):btaf131. doi: 10.1093/bioinformatics/btaf131. Bioinformatics. 2025. PMID: 40139667 Free PMC article.
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
