

DaXi-high-resolution, large imaging volume and multi-view single-objective light-sheet microscopy
- PMID: 35314838
- PMCID: PMC9007742
- DOI: 10.1038/s41592-022-01417-2
DaXi-high-resolution, large imaging volume and multi-view single-objective light-sheet microscopy
Abstract
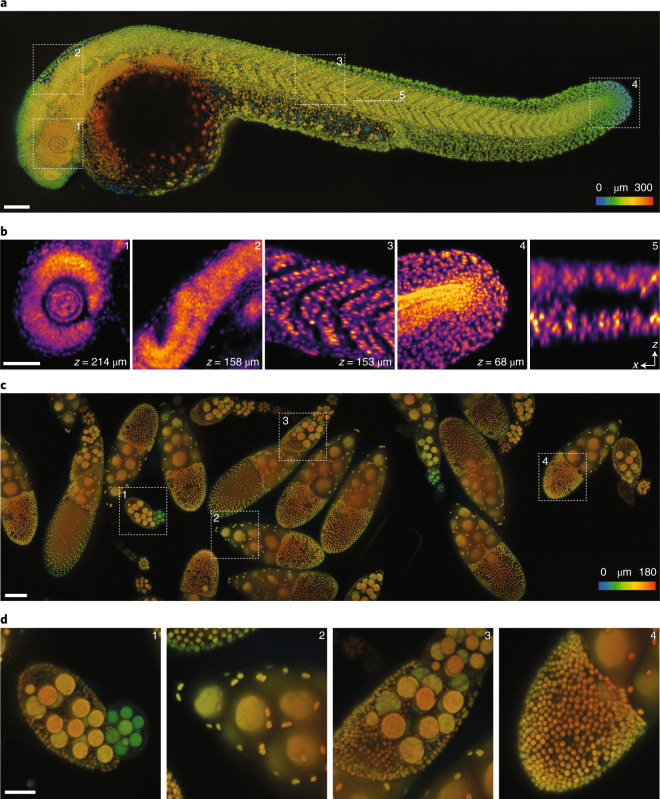
The promise of single-objective light-sheet microscopy is to combine the convenience of standard single-objective microscopes with the speed, coverage, resolution and gentleness of light-sheet microscopes. We present DaXi, a single-objective light-sheet microscope design based on oblique plane illumination that achieves: (1) a wider field of view and high-resolution imaging via a custom remote focusing objective; (2) fast volumetric imaging over larger volumes without compromising image quality or necessitating tiled acquisition; (3) fuller image coverage for large samples via multi-view imaging and (4) higher throughput multi-well imaging via remote coverslip placement. Our instrument achieves a resolution of 450 nm laterally and 2 μm axially over an imaging volume of 3,000 × 800 × 300 μm. We demonstrate the speed, field of view, resolution and versatility of our instrument by imaging various systems, including Drosophila egg chamber development, zebrafish whole-brain activity and zebrafish embryonic development - up to nine embryos at a time.
© 2022. The Author(s).
Conflict of interest statement
A patent application has been filed by B.Y. and L.A.R. covering the reported microscope design. The remaining authors declare no competing interests.
Figures














References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
