

Review article: Lynch Syndrome-a mechanistic and clinical management update
- PMID: 35315099
- PMCID: PMC10936860
- DOI: 10.1111/apt.16826
Review article: Lynch Syndrome-a mechanistic and clinical management update
Abstract
Background: Lynch syndrome (LS) is an autosomal dominant familial condition caused by a pathogenic variant (PV) in a DNA mismatch repair gene, which then predisposes carriers to various cancers.
Aim: To review the pathogenesis, clinical presentation, differential diagnosis and clinical strategies for detection and management of LS.
Methods: A narrative review synthesising knowledge from published literature, as well as current National Comprehensive Cancer Network guidelines for management of LS was conducted.
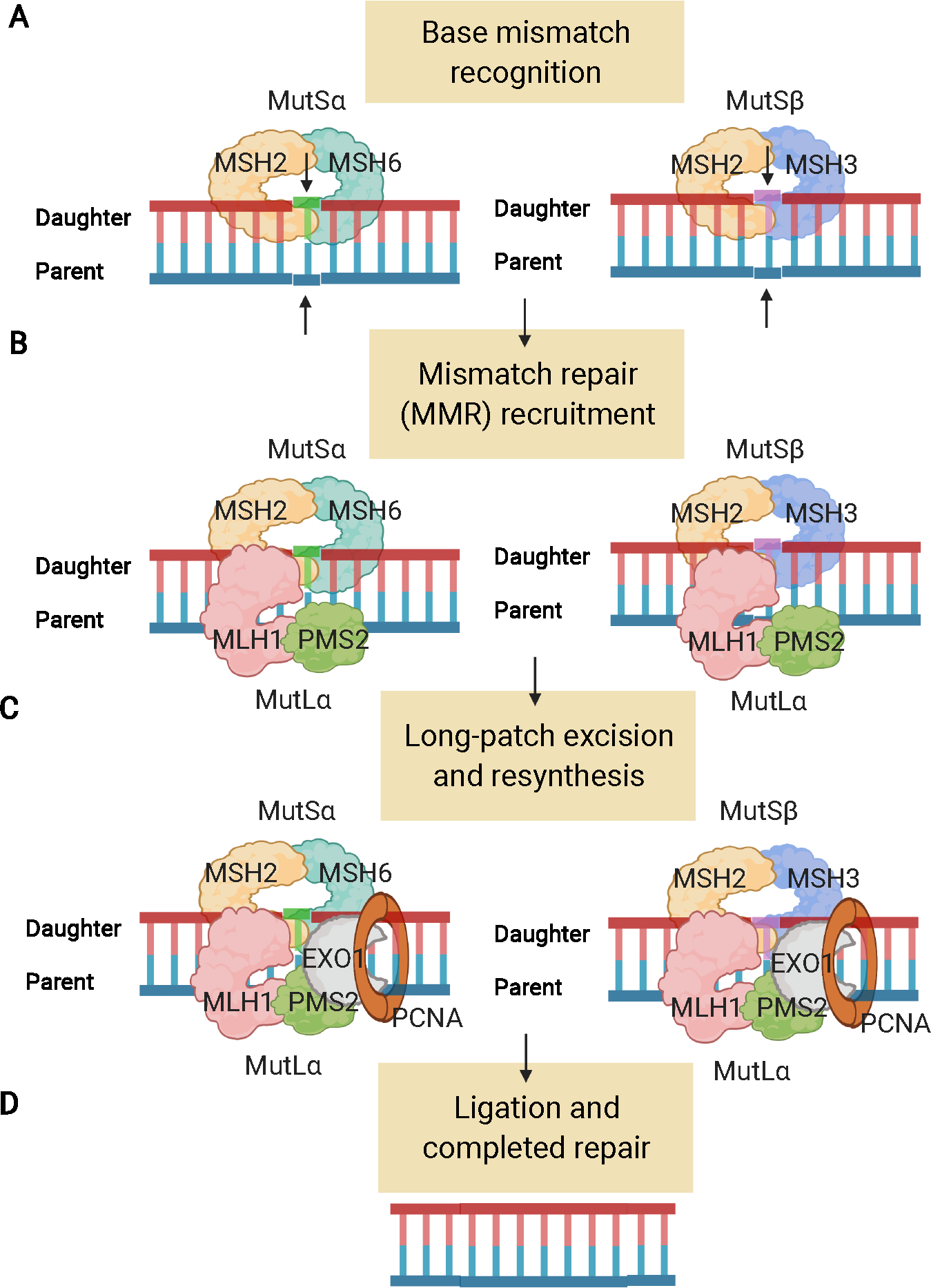
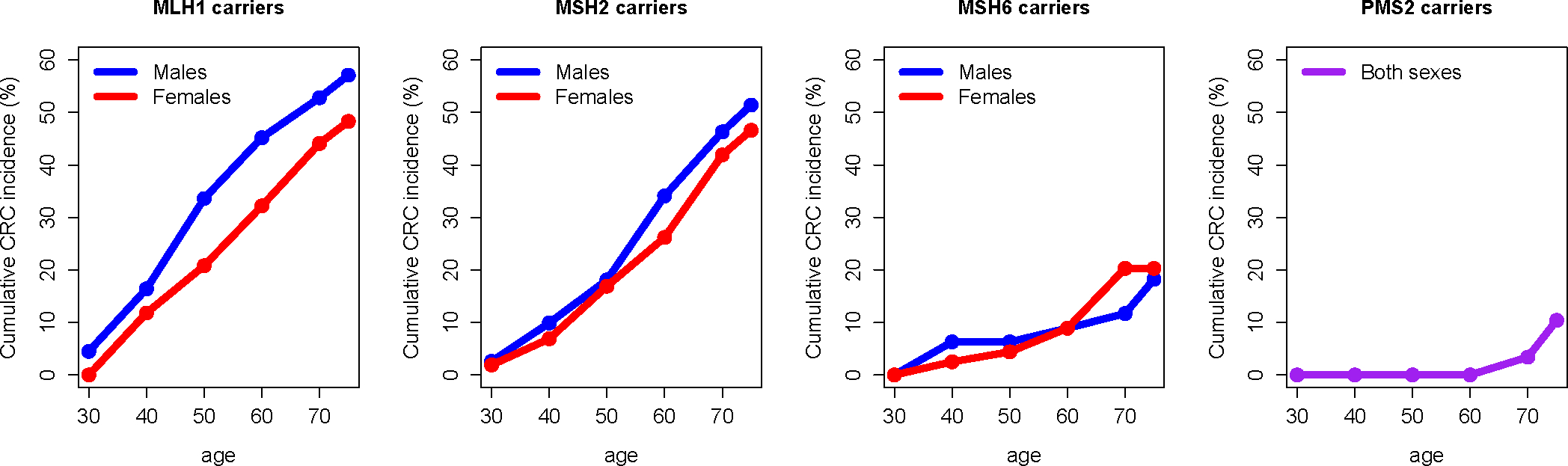
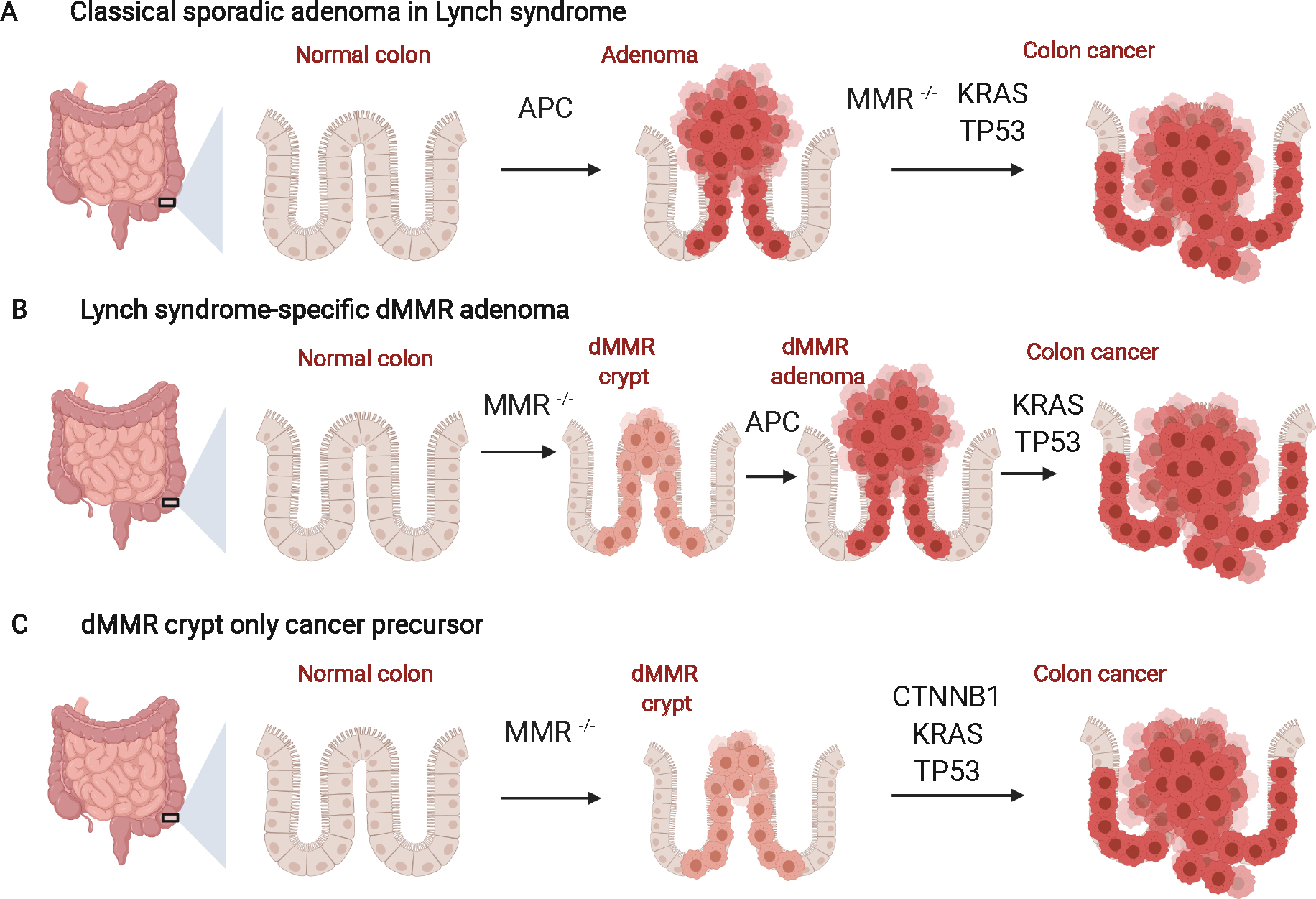
Results: LS tumours are characterised by unique pathogenesis, ultimately resulting in hypermutation, microsatellite instability and high immunogenicity that has significant implications for cancer risk, clinical presentation, treatment and surveillance. LS is one of the most common hereditary causes of cancer, and about 1 in 279 individuals carry a PV in an LS gene that predisposes to associated cancers. Individuals with LS have increased risks for colorectal, endometrial and other cancers, with significant variation in lifetime risk by LS-associated gene.
Conclusions: As genetic testing becomes more widespread, the number of individuals identified with LS is expected to increase in the population. Understanding the pathogenesis of LS informs current strategies for detection and clinical management, and also guides future areas for clinical innovation. Unravelling the mechanisms by which these tumours evolve may help to more precisely tailor management by the gene involved.
© 2022 John Wiley & Sons Ltd.
Figures
References
-
- Win AK, Jenkins MA, Dowty JG, et al. Prevalence and Penetrance of Major Genes and Polygenes for Colorectal Cancer. Cancer epidemiology, biomarkers & prevention : a publication of the American Association for Cancer Research, cosponsored by the American Society of Preventive Oncology. Mar 2017;26(3):404–412. - PMC - PubMed
-
- Peltomaki P. Lynch syndrome genes. Familial cancer. 2005;4(3):227–232. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials