

Consequences of excessive glucosylsphingosine in glucocerebrosidase-deficient zebrafish
- PMID: 35315333
- PMCID: PMC9058576
- DOI: 10.1016/j.jlr.2022.100199
Consequences of excessive glucosylsphingosine in glucocerebrosidase-deficient zebrafish
Abstract
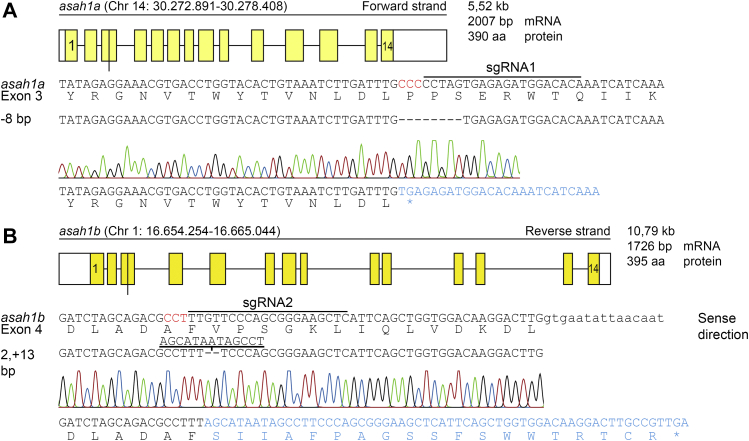
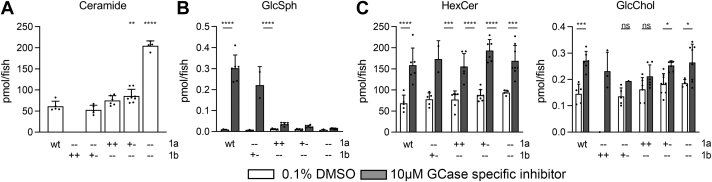
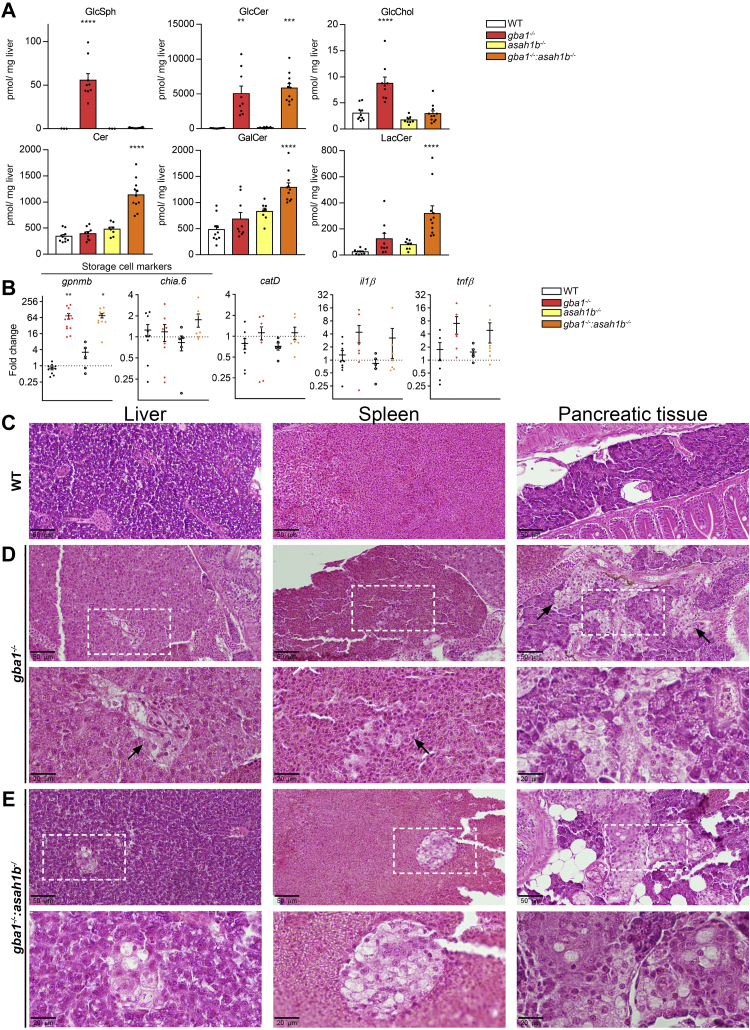
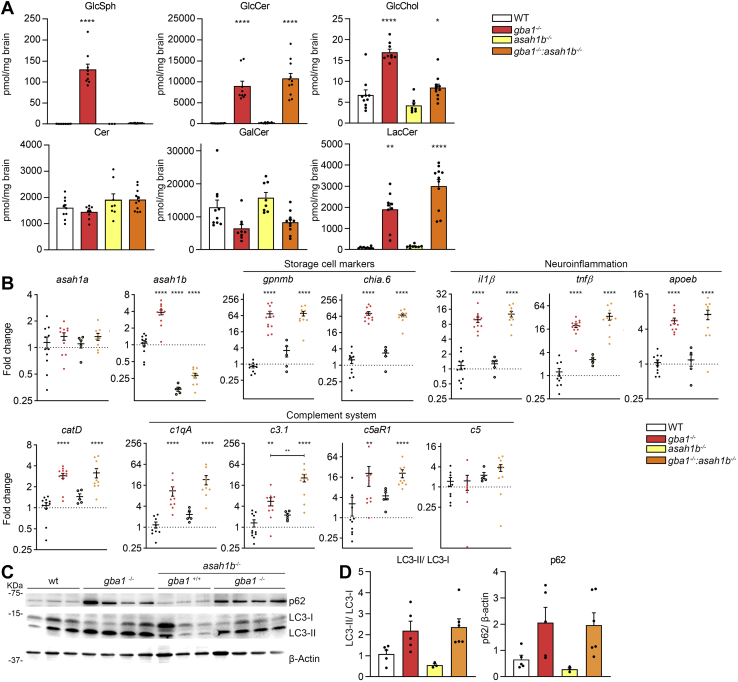
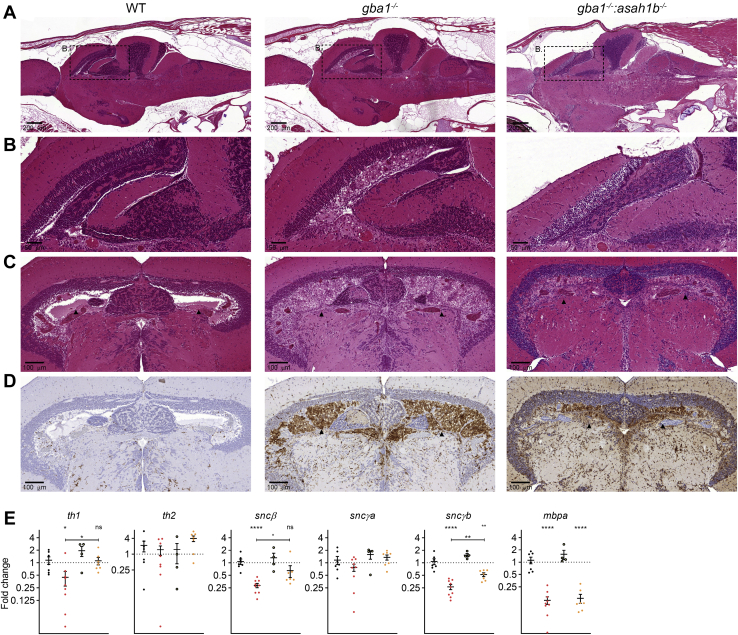
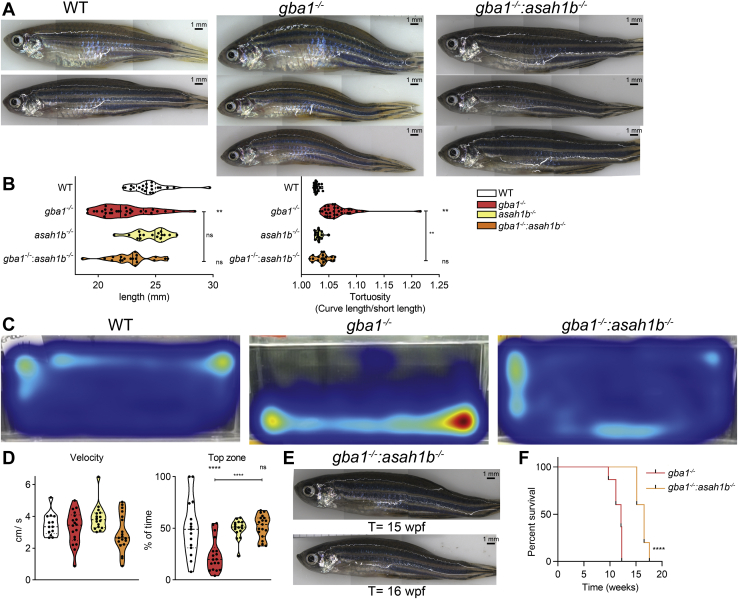
In Gaucher disease (GD), the deficiency of glucocerebrosidase causes lysosomal accumulation of glucosylceramide (GlcCer), which is partly converted by acid ceramidase to glucosylsphingosine (GlcSph) in the lysosome. Chronically elevated blood and tissue GlcSph is thought to contribute to symptoms in GD patients as well as to increased risk for Parkinson's disease. On the other hand, formation of GlcSph may be beneficial since the water soluble sphingoid base is excreted via urine and bile. To study the role of excessive GlcSph formation during glucocerebrosidase deficiency, we studied zebrafish that have two orthologs of acid ceramidase, Asah1a and Asah1b. Only the latter is involved in the formation of GlcSph in glucocerebrosidase-deficient zebrafish as revealed by knockouts of Asah1a or Asah1b with glucocerebrosidase deficiency (either pharmacologically induced or genetic). Comparison of zebrafish with excessive GlcSph (gba1-/- fish) and without GlcSph (gba1-/-:asah1b-/- fish) allowed us to study the consequences of chronic high levels of GlcSph. Prevention of excessive GlcSph in gba1-/-:asah1b-/- fish did not restrict storage cells, GlcCer accumulation, or neuroinflammation. However, GD fish lacking excessive GlcSph show an ameliorated course of disease reflected by significantly increased lifespan, delayed locomotor abnormality, and delayed development of an abnormal curved back posture. The loss of tyrosine hydroxylase 1 (th1) mRNA, a marker of dopaminergic neurons, is slowed down in brain of GD fish lacking excessive GlcSph. In conclusion, in the zebrafish GD model, excess GlcSph has little impact on (neuro)inflammation or the presence of GlcCer-laden macrophages but rather seems harmful to th1-positive dopaminergic neurons.
Keywords: Gaucher disease; acid ceramidase; dopaminergic neurons; glucosylceramide; lipid metabolism; lysosphingolipids; sphingolipids; zebrafish.
Copyright © 2022 The Authors. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Conflict of interest The authors declare that they have no conflicts of interest with the contents of this article.
Figures

References
-
- Ferraz M.J., Kallemeijn W.W., Mirzaian M., Herrera Moro D., Marques A., Wisse P., Boot R.G., Willems L.I., Overkleeft H.S., Aerts J.M. Gaucher disease and Fabry disease: new markers and insights in pathophysiology for two distinct glycosphingolipidoses. Biochim. Biophys. Acta. 2014;1841:811–825. - PubMed
-
- Bodamer O.A., Hung C. Laboratory and genetic evaluation of Gaucher disease. Wien Med. Wochenschr. 2010;160:600–604. - PubMed
-
- Mechtler T.P., Stary S., Metz T.F., De Jesus V.R., Greber-Platzer S., Pollak A., Herkner K.R., Streubel B., Kasper D.C. Neonatal screening for lysosomal storage disorders: feasibility and incidence from a nationwide study in Austria. Lancet. 2012;379:335–341. - PubMed
-
- Pollak A., Kasper D.C. Austrian newborn screening program: a perspective of five decades. J. Perinat. Med. 2014;42:151–158. - PubMed
-
- Platt F.M., d'Azzo A., Davidson B.L., Neufeld E.F., Tifft C.J. Lysosomal storage diseases. Nat. Rev. Dis. Primers. 2018;4:27. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
