

Global population structure of the Serratia marcescens complex and identification of hospital-adapted lineages in the complex
- PMID: 35315751
- PMCID: PMC9176281
- DOI: 10.1099/mgen.0.000793
Global population structure of the Serratia marcescens complex and identification of hospital-adapted lineages in the complex
Abstract
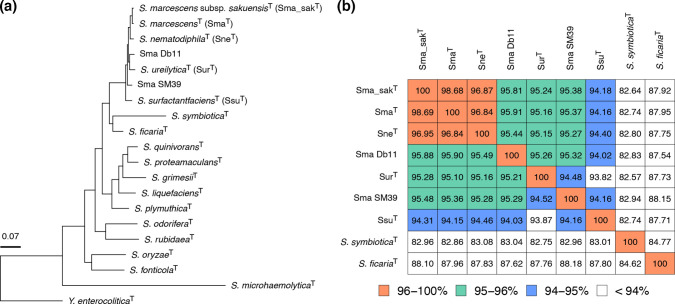
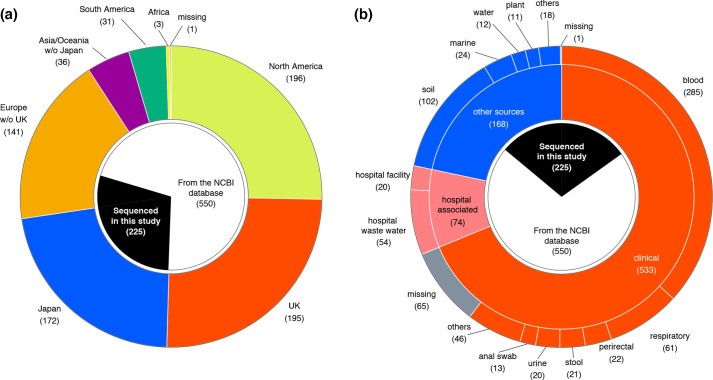
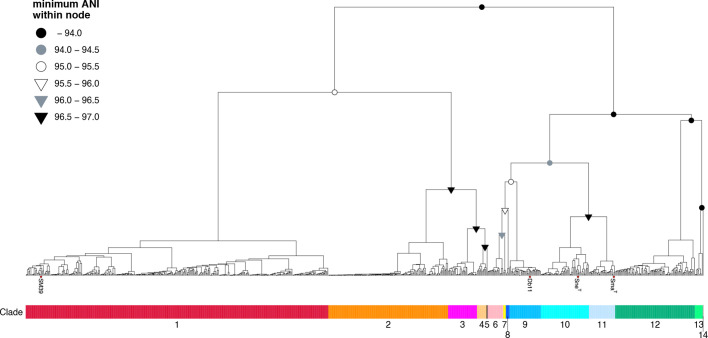
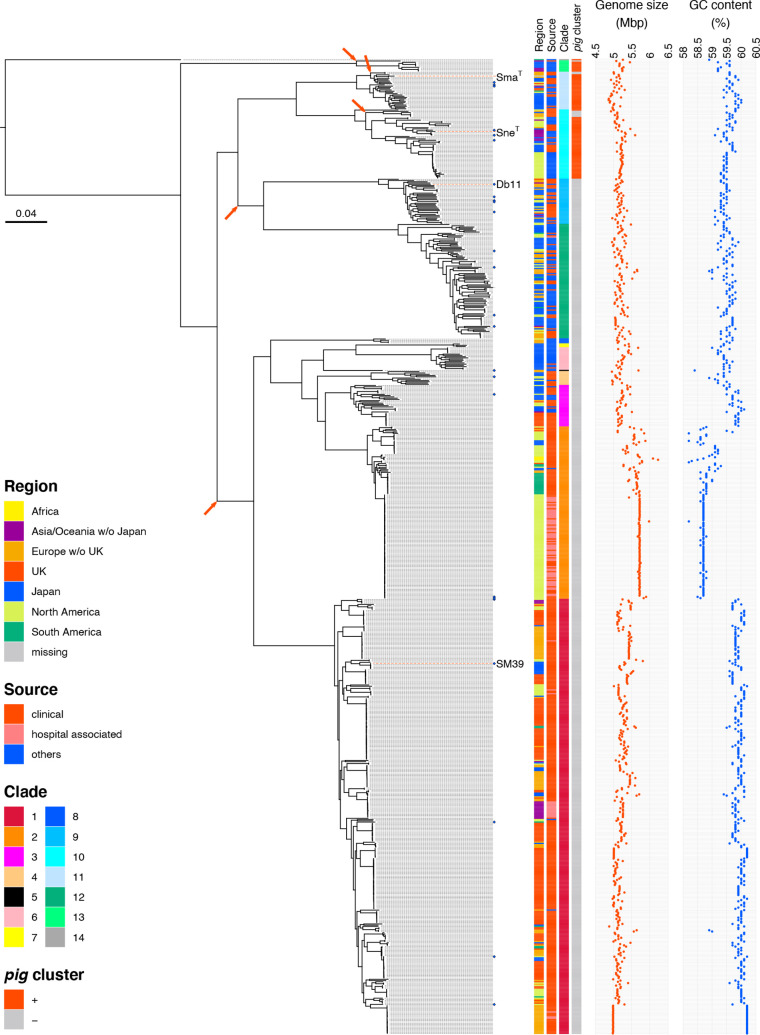
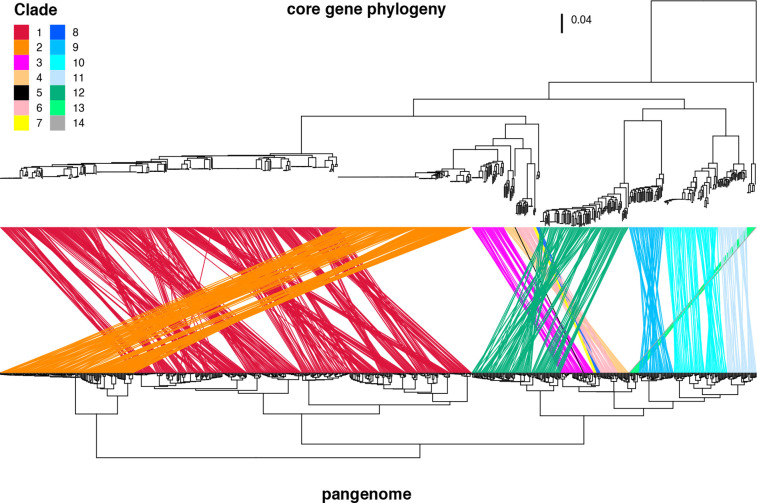
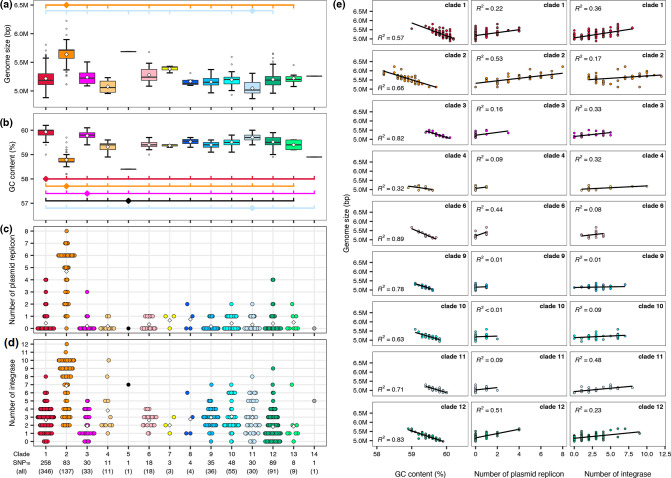
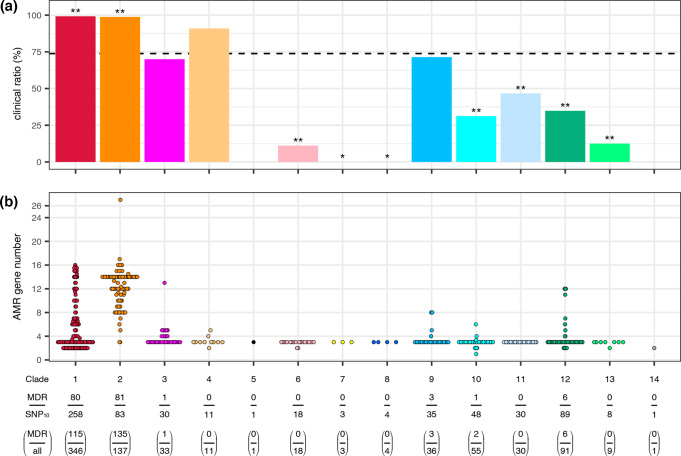
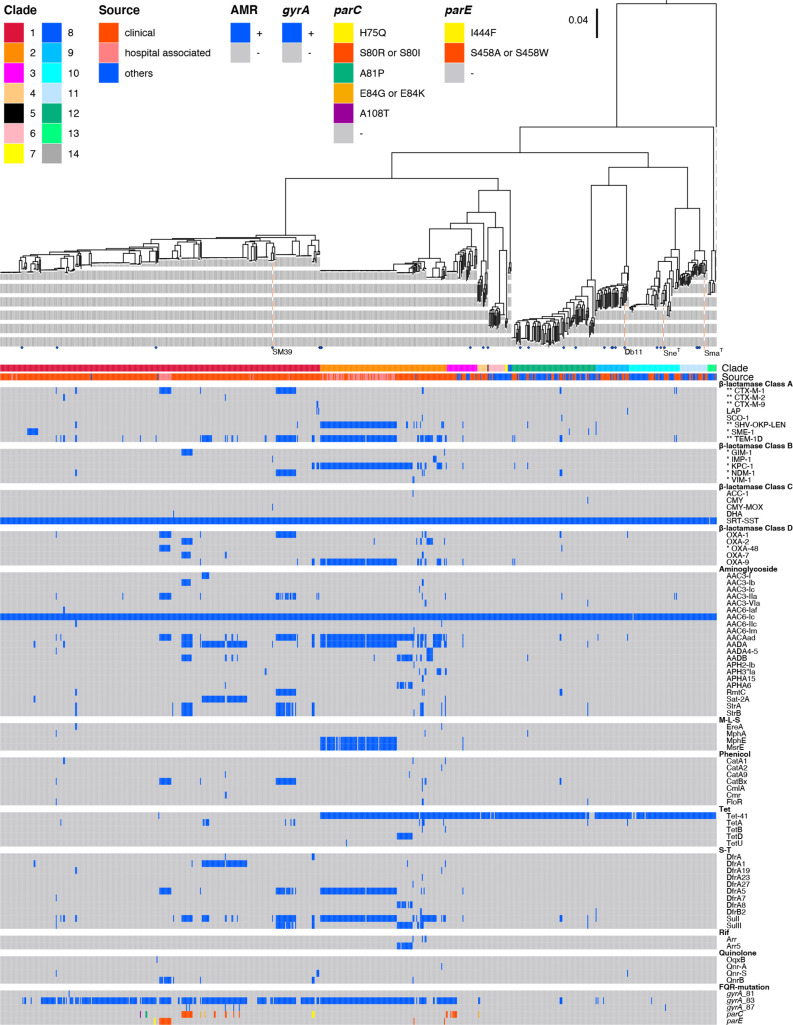
Serratia marcescens is an important nosocomial pathogen causing various opportunistic infections, such as urinary tract infections, bacteremia and sometimes even hospital outbreaks. The recent emergence and spread of multidrug-resistant (MDR) strains further pose serious threats to global public health. This bacterium is also ubiquitously found in natural environments, but the genomic differences between clinical and environmental isolates are not clear, including those between S. marcescens and its close relatives. In this study, we performed a large-scale genome analysis of S. marcescens and closely related species (referred to as the 'S. marcescens complex'), including more than 200 clinical and environmental strains newly sequenced here. Our analysis revealed their phylogenetic relationships and complex global population structure, comprising 14 clades, which were defined based on whole-genome average nucleotide identity. Clades 10, 11, 12 and 13 corresponded to S. nematodiphila, S. marcescens sensu stricto, S. ureilytica and S. surfactantfaciens, respectively. Several clades exhibited distinct genome sizes and GC contents and a negative correlation of these genomic parameters was observed in each clade, which was associated with the acquisition of mobile genetic elements (MGEs), but different types of MGEs, plasmids or prophages (and other integrative elements), were found to contribute to the generation of these genomic variations. Importantly, clades 1 and 2 mostly comprised clinical or hospital environment isolates and accumulated a wide range of antimicrobial resistance genes, including various extended-spectrum β-lactamase and carbapenemase genes, and fluoroquinolone target site mutations, leading to a high proportion of MDR strains. This finding suggests that clades 1 and 2 represent hospital-adapted lineages in the S. marcescens complex although their potential virulence is currently unknown. These data provide an important genomic basis for reconsidering the classification of this group of bacteria and reveal novel insights into their evolution, biology and differential importance in clinical settings.
Keywords: Serratia marcescens complex; antimicrobial resistance; genomics; hospital-adapted lineage; population structure.
Conflict of interest statement
The authors declare that there are no conflicts of interest.
Figures
References
-
- Herbert S, Halvorsen DS, Leong T, Franklin C, Harrington G, et al. Large outbreak of infection and colonization with gram-negative pathogens carrying the metallo- beta -lactamase gene blaIMP-4 at a 320-bed tertiary hospital in Australia. Infect Control Hosp Epidemiol. 2007;28:98–101. doi: 10.1086/508841. - DOI - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous