

Combining genotypes and T cell receptor distributions to infer genetic loci determining V(D)J recombination probabilities
- PMID: 35315770
- PMCID: PMC8940181
- DOI: 10.7554/eLife.73475
Combining genotypes and T cell receptor distributions to infer genetic loci determining V(D)J recombination probabilities
Abstract
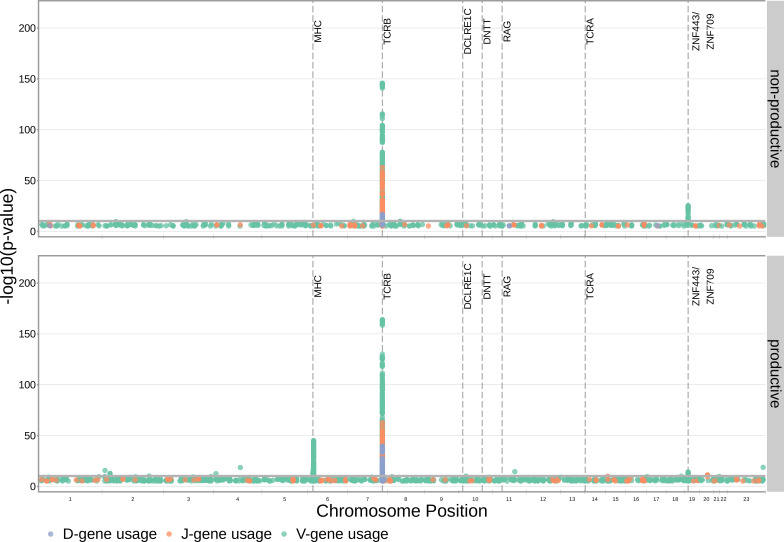
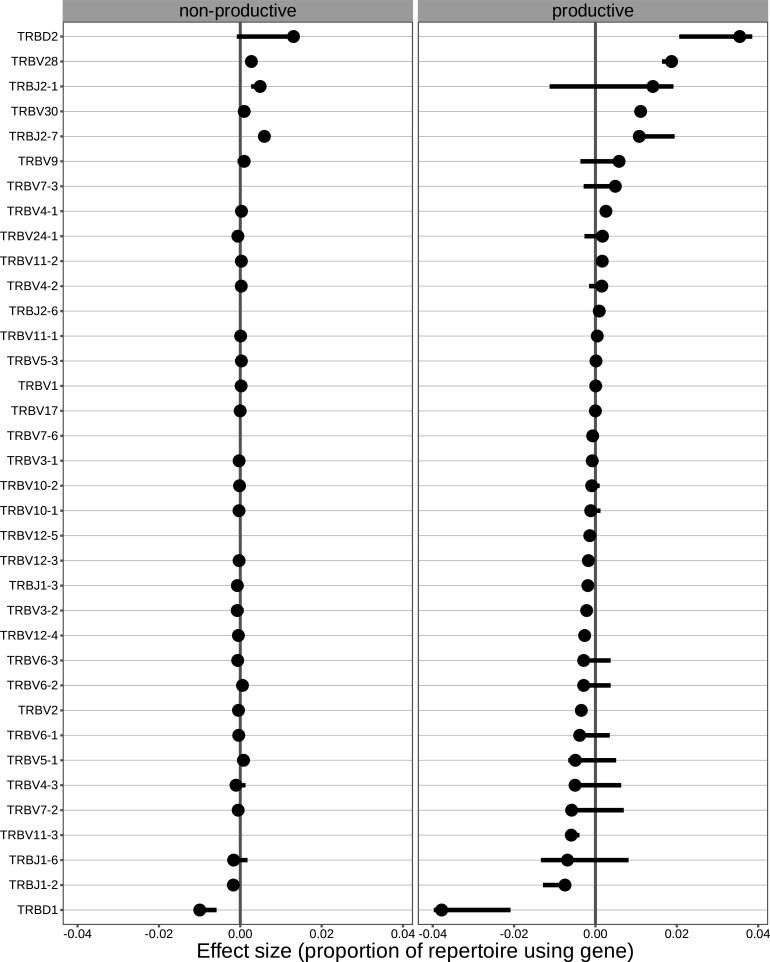
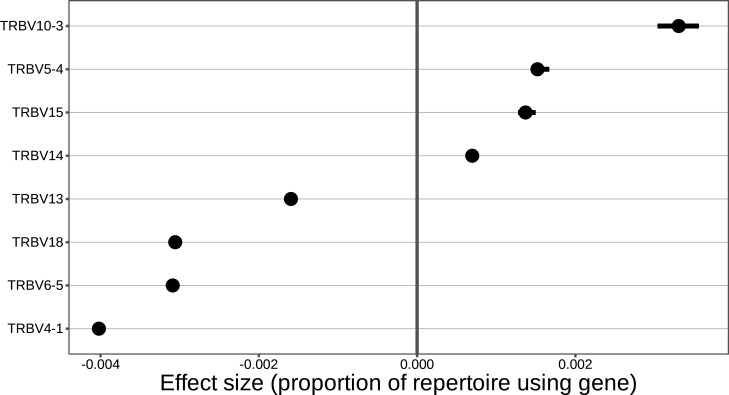
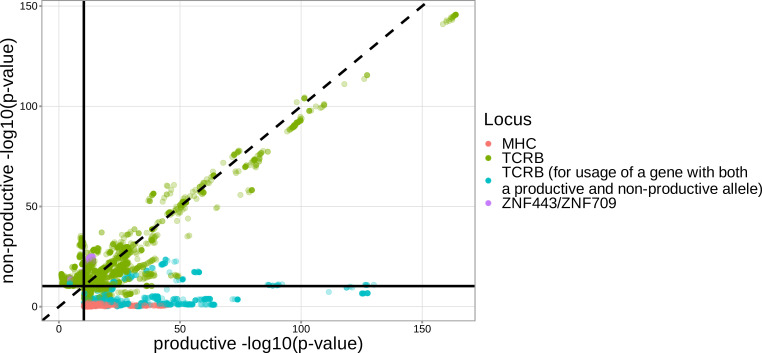
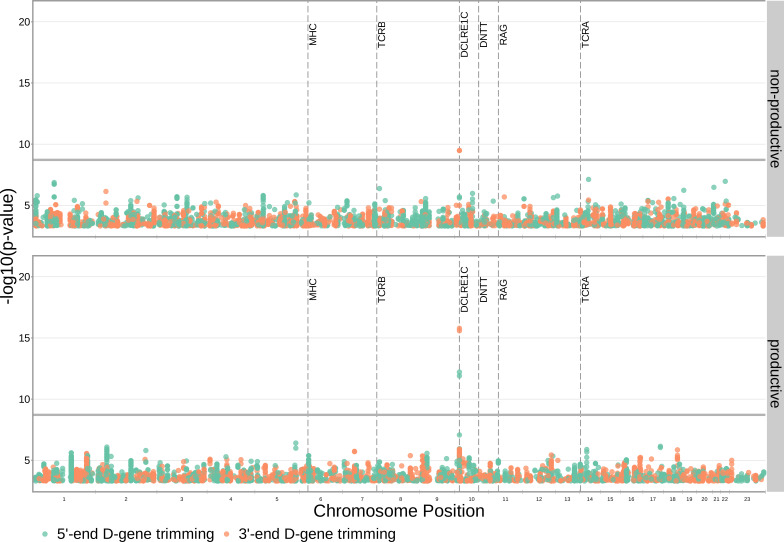
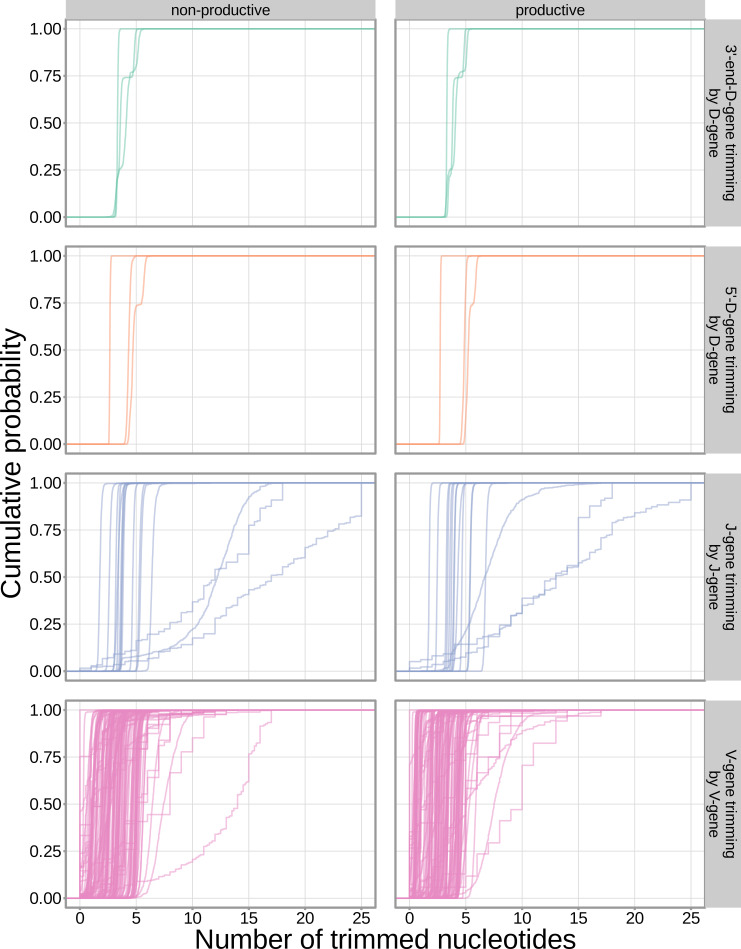
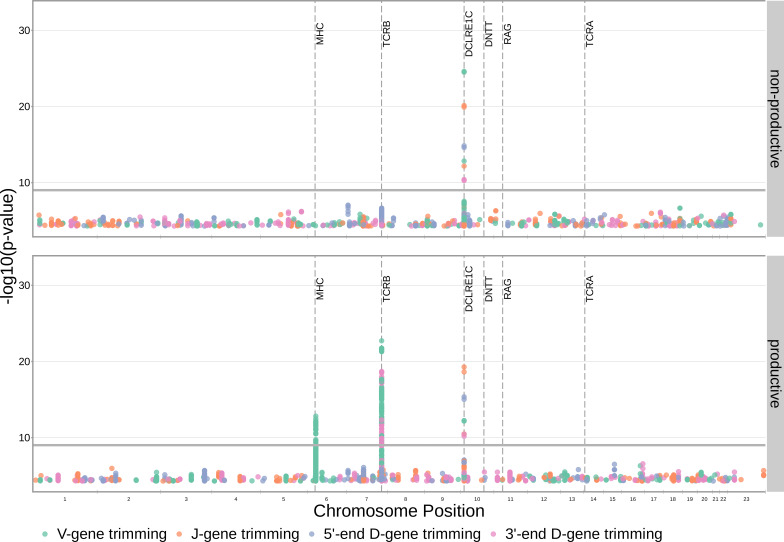
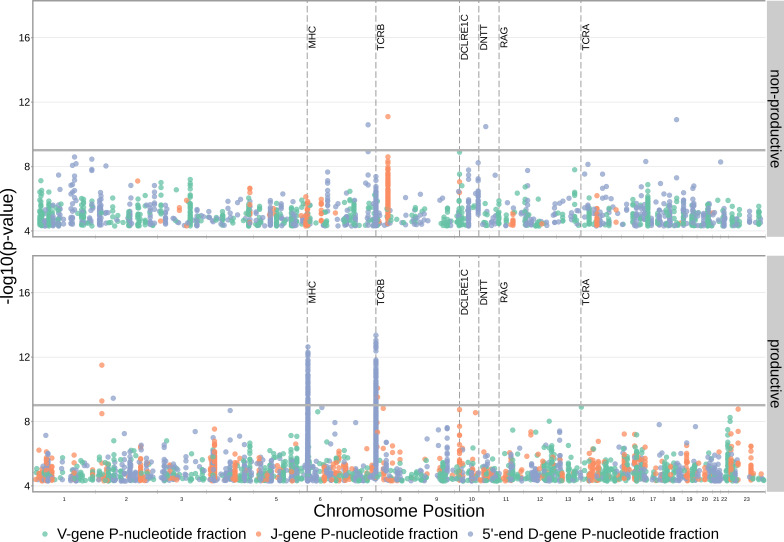
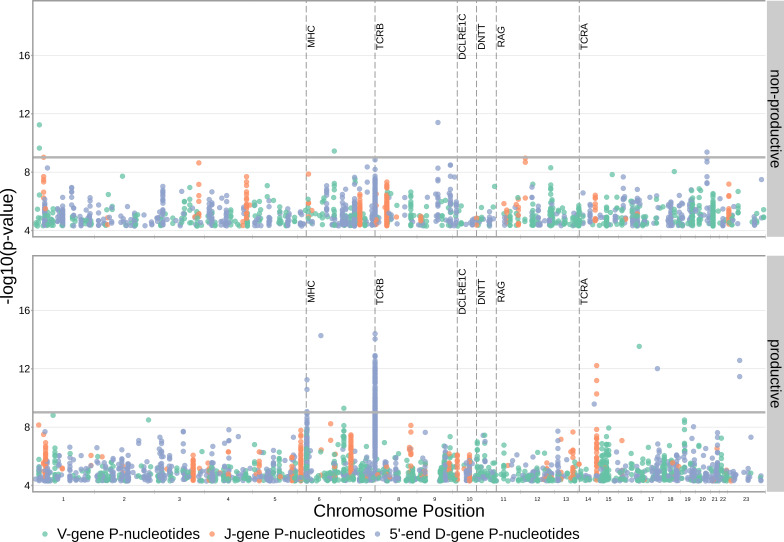
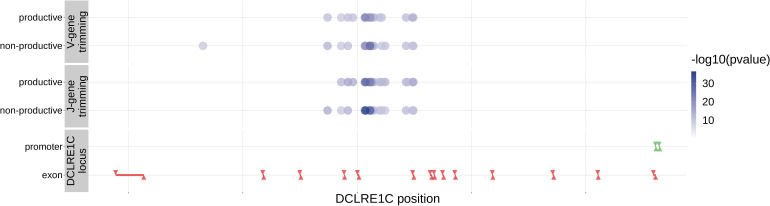
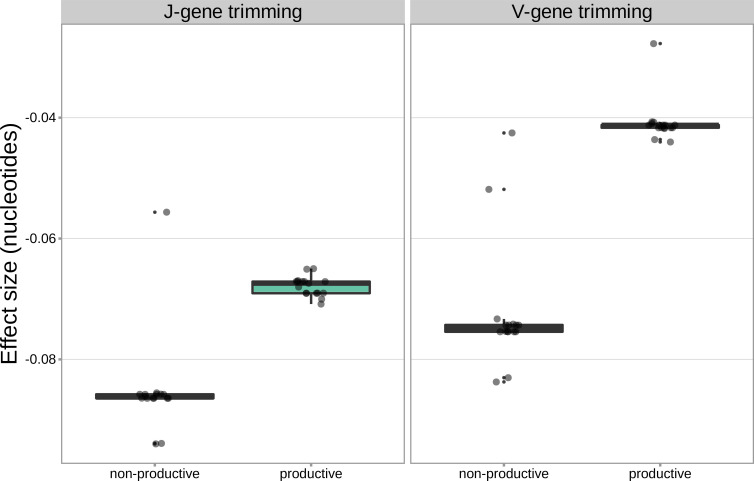
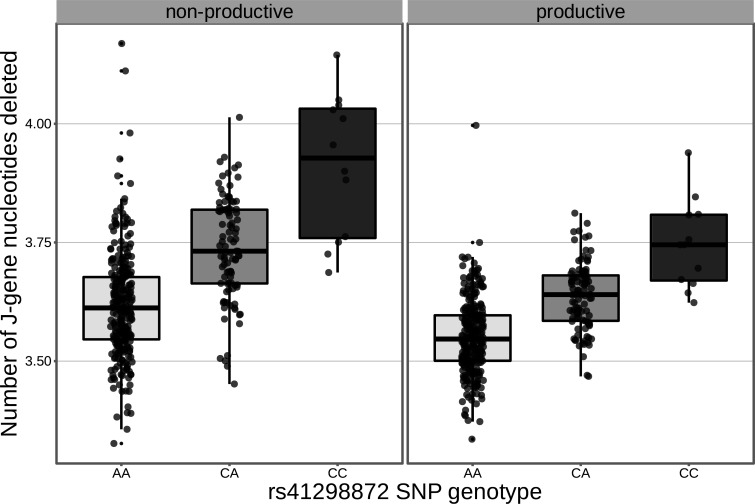
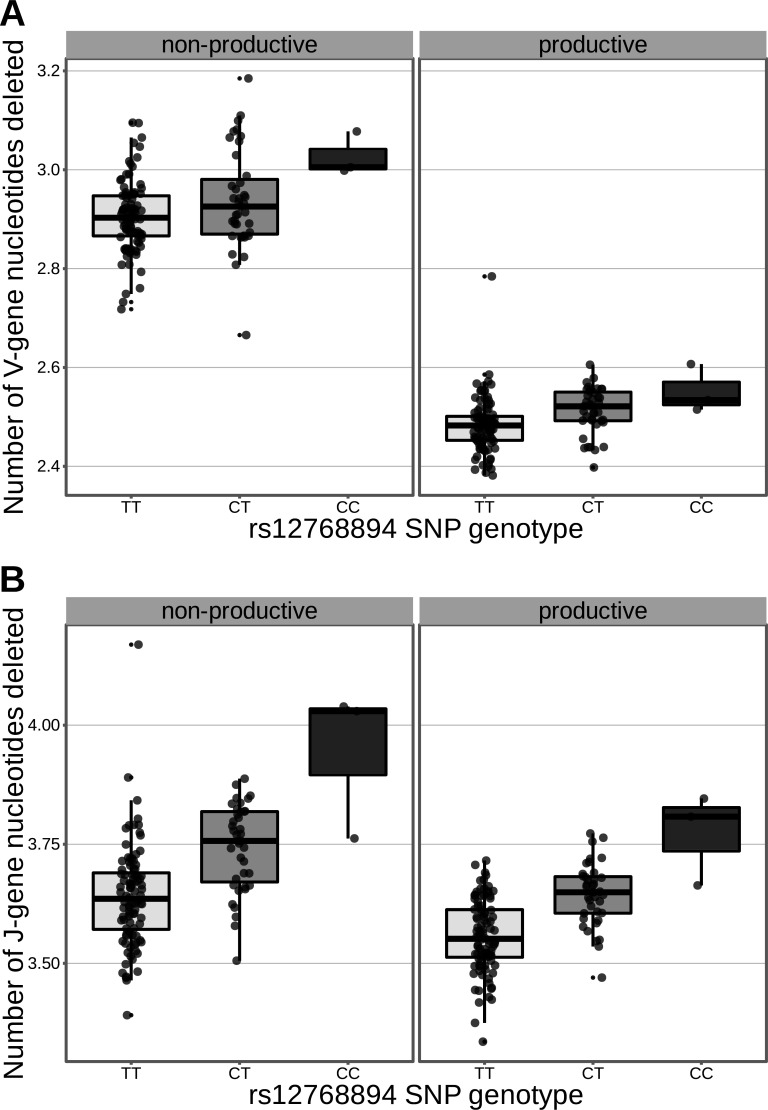
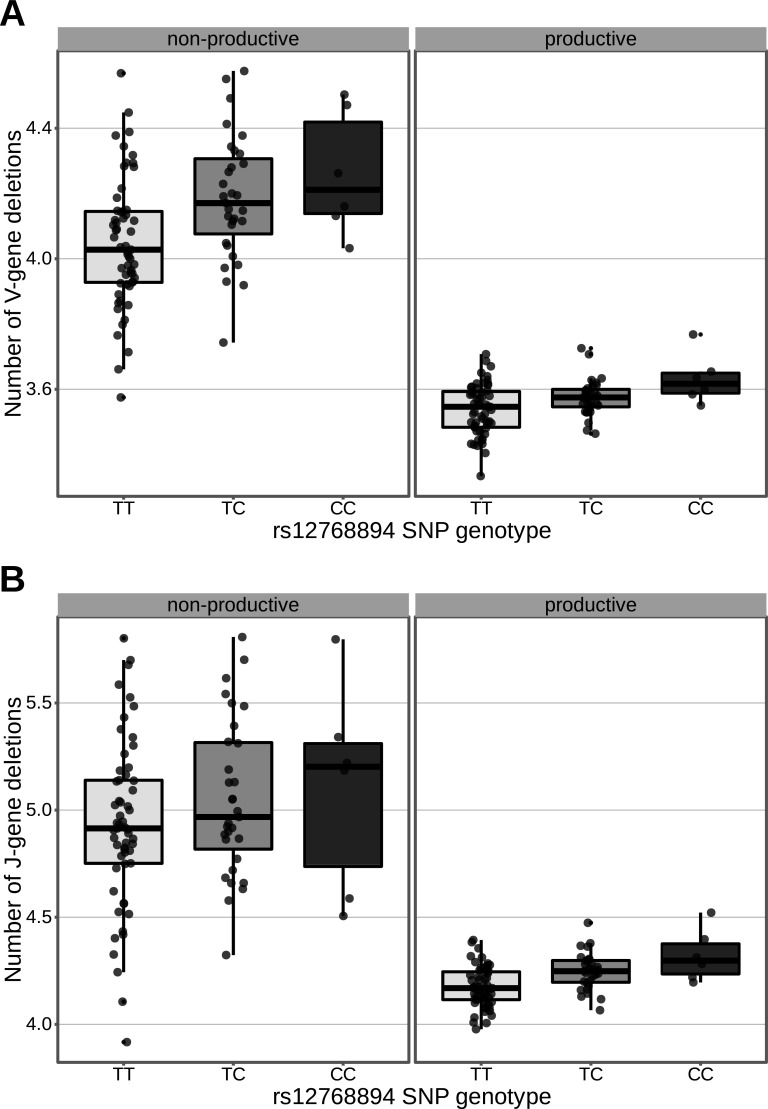
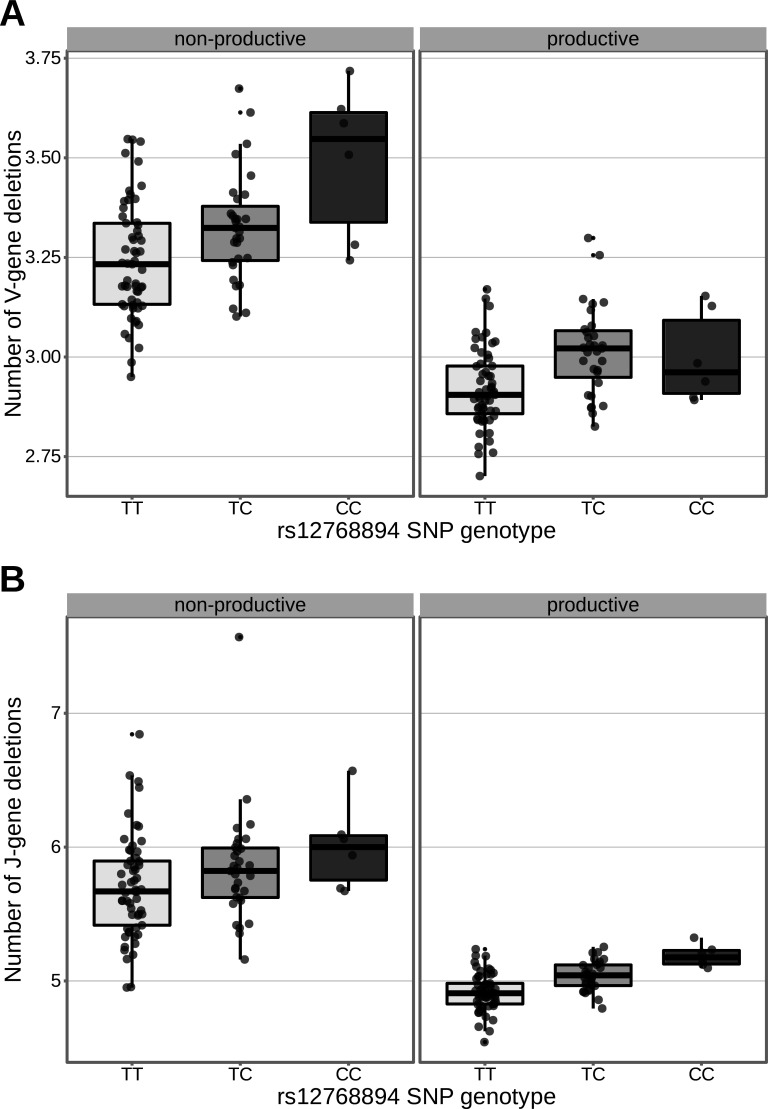
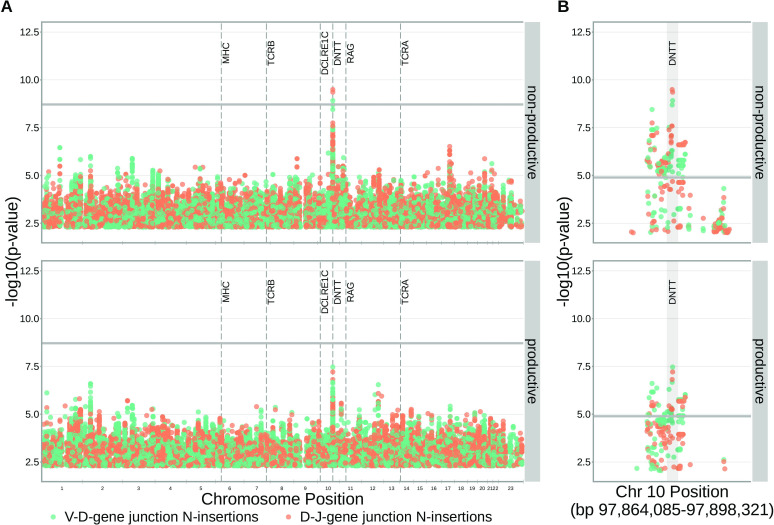
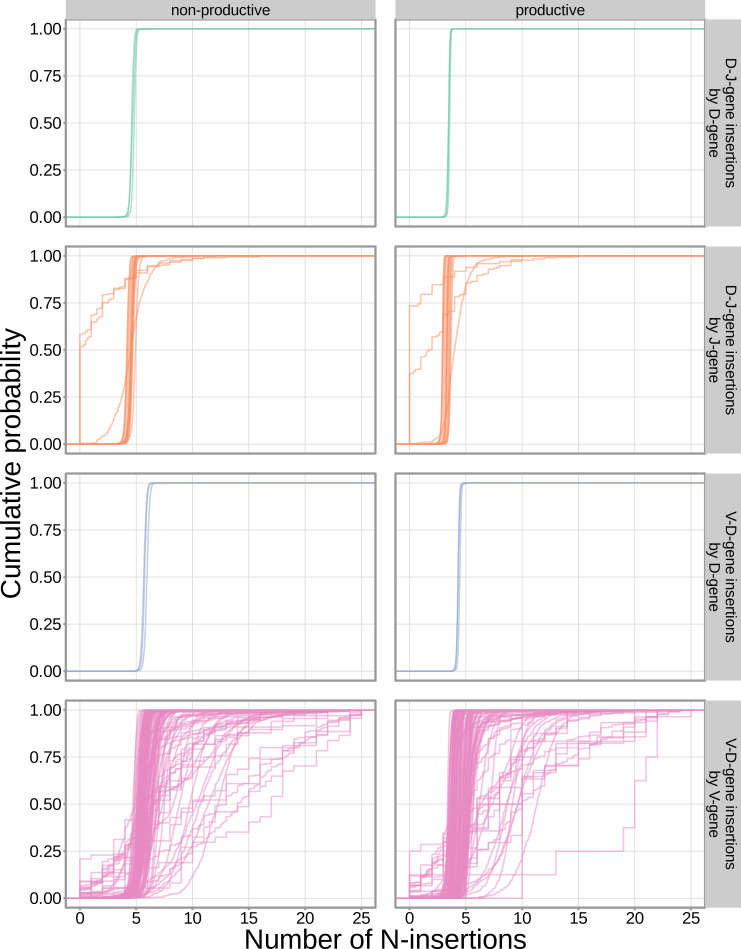
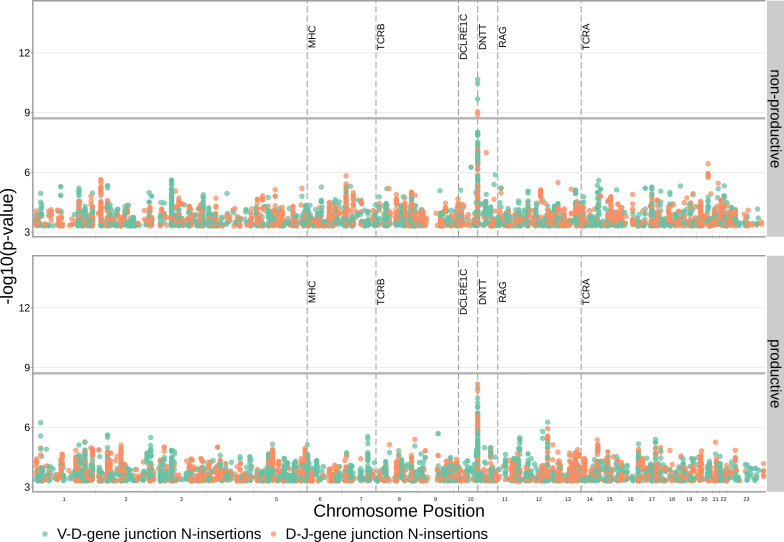
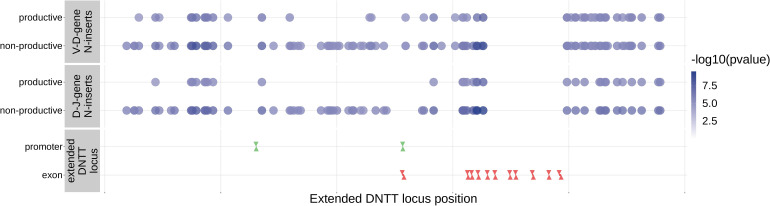
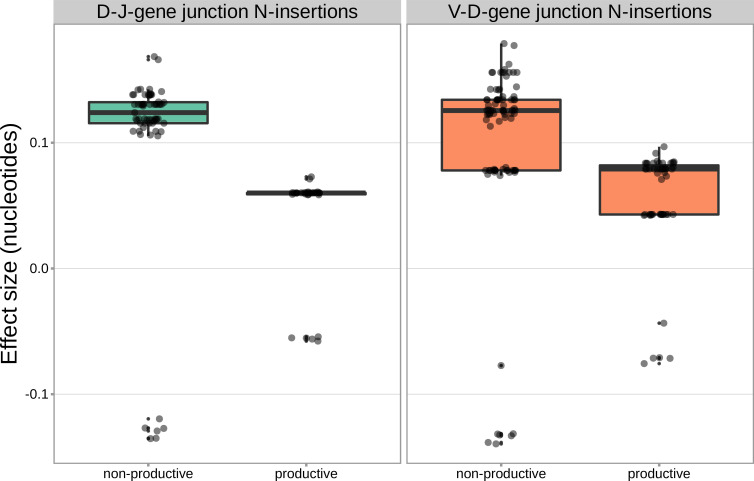
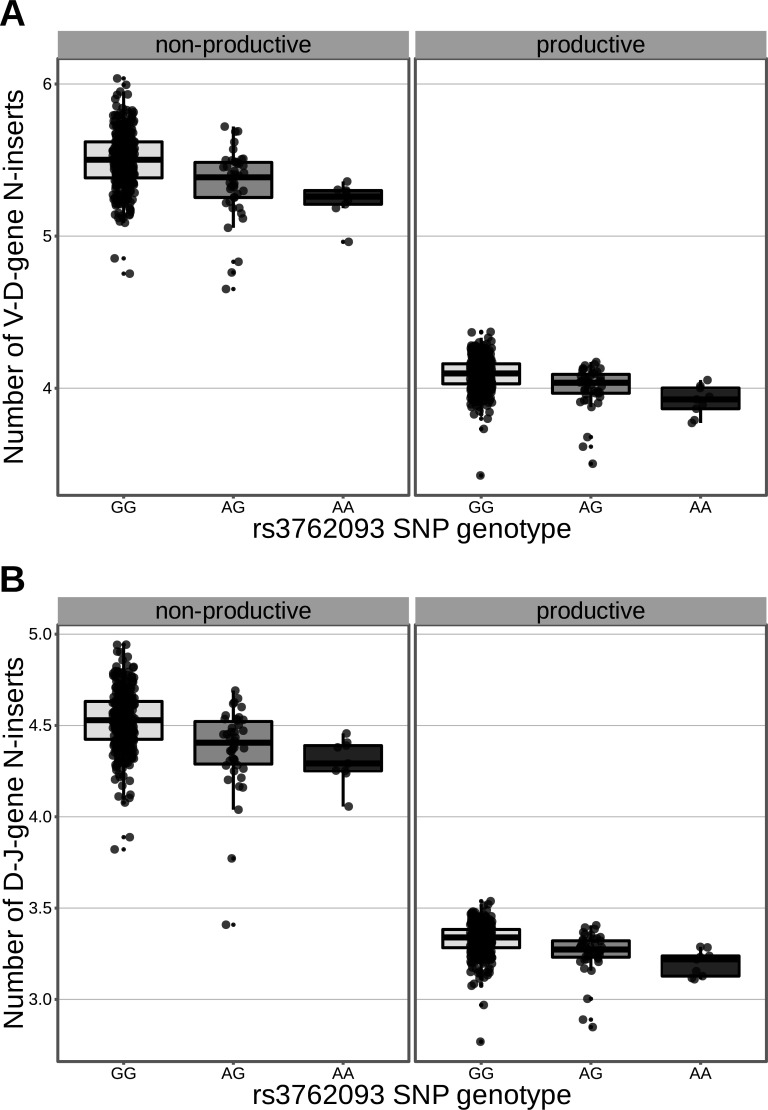
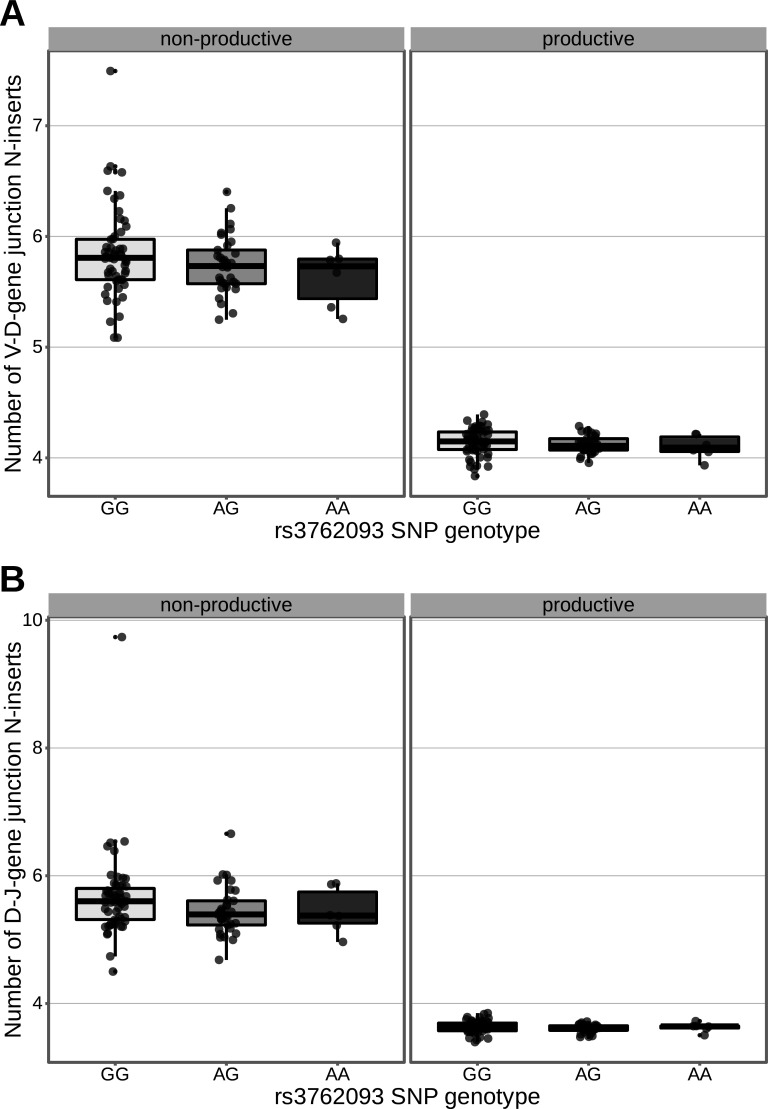
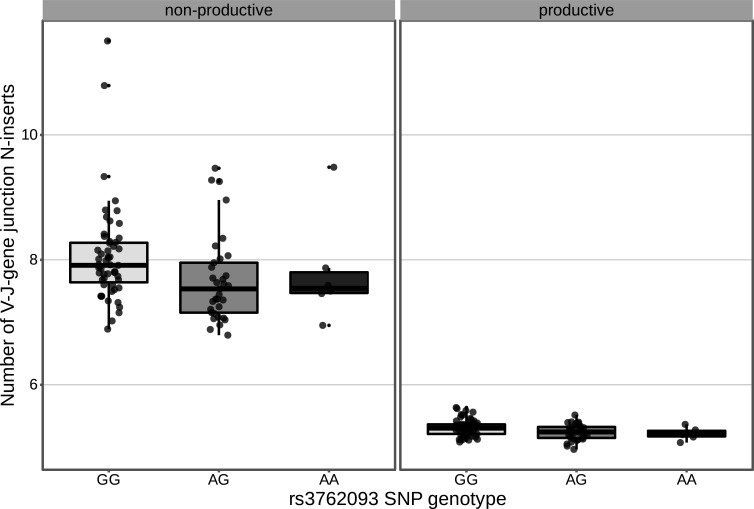
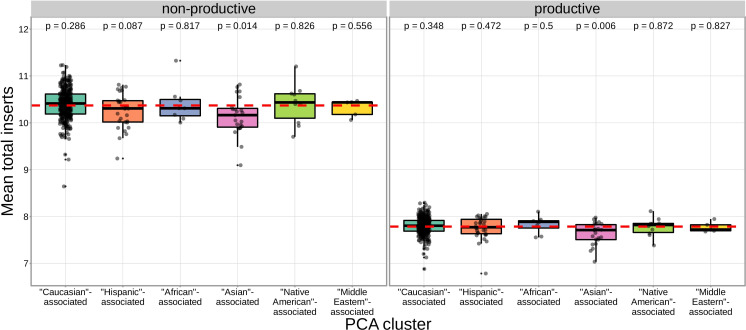
Every T cell receptor (TCR) repertoire is shaped by a complex probabilistic tangle of genetically determined biases and immune exposures. T cells combine a random V(D)J recombination process with a selection process to generate highly diverse and functional TCRs. The extent to which an individual's genetic background is associated with their resulting TCR repertoire diversity has yet to be fully explored. Using a previously published repertoire sequencing dataset paired with high-resolution genome-wide genotyping from a large human cohort, we infer specific genetic loci associated with V(D)J recombination probabilities using genome-wide association inference. We show that V(D)J gene usage profiles are associated with variation in the TCRB locus and, specifically for the functional TCR repertoire, variation in the major histocompatibility complex locus. Further, we identify specific variations in the genes encoding the Artemis protein and the TdT protein to be associated with biasing junctional nucleotide deletion and N-insertion, respectively. These results refine our understanding of genetically-determined TCR repertoire biases by confirming and extending previous studies on the genetic determinants of V(D)J gene usage and providing the first examples of trans genetic variants which are associated with modifying junctional diversity. Together, these insights lay the groundwork for further explorations into how immune responses vary between individuals.
Keywords: Artemis; GWAS; TdT; VDJ recombination probabilities; human; immunology; inflammation; t cell receptor repertoire.
© 2022, Russell et al.
Conflict of interest statement
MR, AS, DL, SS, EA, GK, NS, AB, FM, PB No competing interests declared, AG serves on a scientific advisory board for Janssen, PT consults for Johnson and Johnson, Immunoscape, Cytoagents, and PACT Pharma. He has received travel reimbursement from 10X Genomics and Illumina. He is an inventor on two pending US patent applications related to T cell receptor biology (US: 15/780,938 titled "Cloning and Expression System for T-Cell Receptors' and US: 17/616,279 titled "Kit and Method for Analyzing Singlet Cells')
Figures
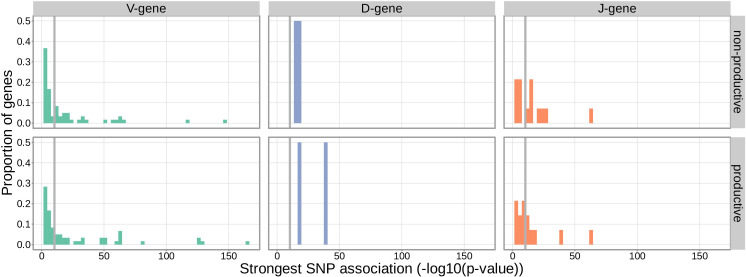
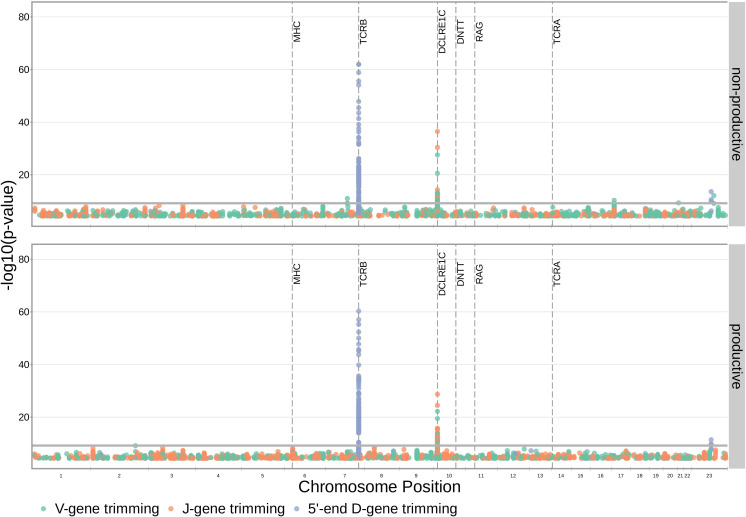
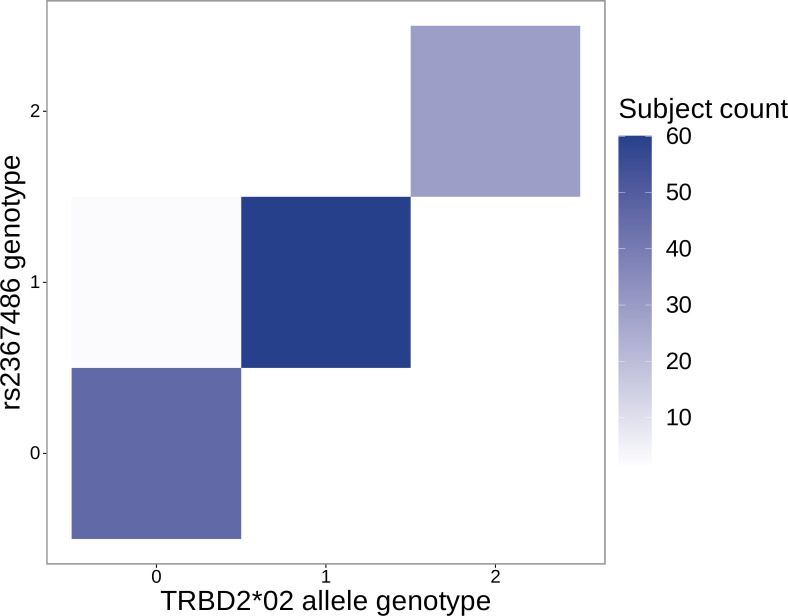
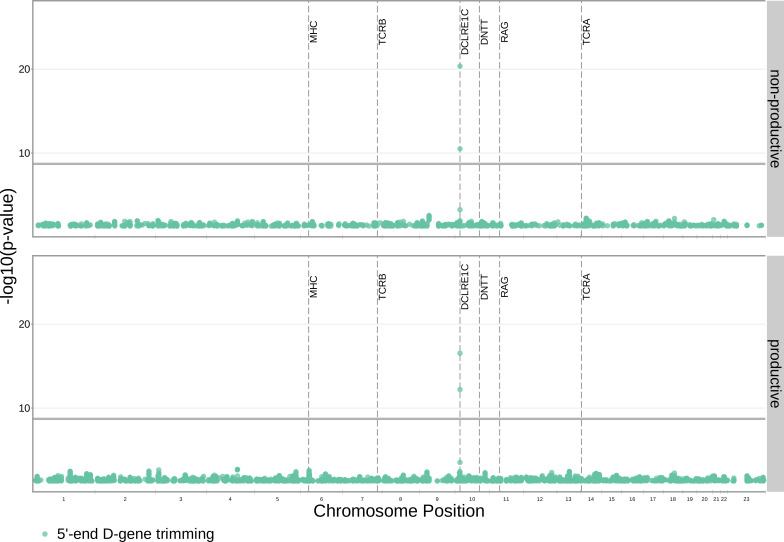
Similar articles
-
Statistical inference reveals the role of length, GC content, and local sequence in V(D)J nucleotide trimming.Elife. 2023 May 25;12:e85145. doi: 10.7554/eLife.85145. Elife. 2023. PMID: 37227256 Free PMC article.
-
XLF deficiency results in reduced N-nucleotide addition during V(D)J recombination.Blood. 2016 Aug 4;128(5):650-9. doi: 10.1182/blood-2016-02-701029. Epub 2016 Jun 8. Blood. 2016. PMID: 27281794 Free PMC article.
-
A Framework for Annotation of Antigen Specificities in High-Throughput T-Cell Repertoire Sequencing Studies.Front Immunol. 2019 Sep 26;10:2159. doi: 10.3389/fimmu.2019.02159. eCollection 2019. Front Immunol. 2019. PMID: 31616409 Free PMC article.
-
RAG Chromatin Scanning During V(D)J Recombination and Chromatin Loop Extrusion are Related Processes.Adv Immunol. 2018;139:93-135. doi: 10.1016/bs.ai.2018.07.001. Epub 2018 Aug 27. Adv Immunol. 2018. PMID: 30249335 Review.
-
Spatial Regulation of V-(D)J Recombination at Antigen Receptor Loci.Adv Immunol. 2015;128:93-121. doi: 10.1016/bs.ai.2015.07.006. Epub 2015 Aug 13. Adv Immunol. 2015. PMID: 26477366 Review.
Cited by
-
Immunosequencing and Profiling of T Cells at the Maternal-Fetal Interface of Women with Preterm Labor and Chronic Chorioamnionitis.J Immunol. 2023 Oct 1;211(7):1082-1098. doi: 10.4049/jimmunol.2300201. J Immunol. 2023. PMID: 37647360 Free PMC article.
-
Interpretable GWAS by linking clinical phenotypes to quantifiable immune repertoire components.Commun Biol. 2024 Oct 20;7(1):1357. doi: 10.1038/s42003-024-07010-x. Commun Biol. 2024. PMID: 39428403 Free PMC article.
-
Targeted long-read sequencing facilitates phased diploid assembly and genotyping of the human T cell receptor alpha, delta, and beta loci.Cell Genom. 2022 Nov 30;2(12):100228. doi: 10.1016/j.xgen.2022.100228. eCollection 2022 Dec 14. Cell Genom. 2022. PMID: 36778049 Free PMC article.
-
Evolutionary trade-offs constraining the MHC gene expansion: beyond simple TCR depletion model.Front Immunol. 2024 Jan 8;14:1240723. doi: 10.3389/fimmu.2023.1240723. eCollection 2023. Front Immunol. 2024. PMID: 38259496 Free PMC article.
-
Robust and prototypical immune responses toward COVID-19 vaccine in First Nations peoples are impacted by comorbidities.Nat Immunol. 2023 Jun;24(6):966-978. doi: 10.1038/s41590-023-01508-y. Epub 2023 May 29. Nat Immunol. 2023. PMID: 37248417 Free PMC article.
References
-
- Bradley P, Crawford JC, Fiore-Gartland A, Perry A, Diez D. TCRdist pipeline. 0.0.2GitHub. 2017 https://github.com/phbradley/tcr-dist
-
- Conomos MP, Laurie CA, Stilp AM, Gogarten SM, McHugh CP, Nelson SC, Sofer T, Fernández-Rhodes L, Justice AE, Graff M, Young KL, Seyerle AA, Avery CL, Taylor KD, Rotter JI, Talavera GA, Daviglus ML, Wassertheil-Smoller S, Schneiderman N, Heiss G, Kaplan RC, Franceschini N, Reiner AP, Shaffer JR, Barr RG, Kerr KF, Browning SR, Browning BL, Weir BS, Avilés-Santa ML, Papanicolaou GJ, Lumley T, Szpiro AA, North KE, Rice K, Thornton TA, Laurie CC. Genetic Diversity and Association Studies in US Hispanic/Latino Populations: Applications in the Hispanic Community Health Study/Study of Latinos. American Journal of Human Genetics. 2016;98:165–184. doi: 10.1016/j.ajhg.2015.12.001. - DOI - PMC - PubMed
-
- Corporation M, Weston S. doParallel: Foreach Parallel Adaptor for the ’parallel’ Package. R package version 1.0.16R Package 2020
-
- Dash P, Fiore-Gartland AJ, Hertz T, Wang GC, Sharma S, Souquette A, Crawford JC, Clemens EB, Nguyen THO, Kedzierska K, La Gruta NL, Bradley P, Thomas PG. Quantifiable predictive features define epitope-specific T cell receptor repertoires. Nature. 2017;547:89–93. doi: 10.1038/nature22383. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Associated data
Grants and funding
- R01 HL094260/HL/NHLBI NIH HHS/United States
- HHSN272201400006C/AI/NIAID NIH HHS/United States
- K23 HL069860/HL/NHLBI NIH HHS/United States
- P01 AI033484/AI/NIAID NIH HHS/United States
- R01 HL105914/HL/NHLBI NIH HHS/United States
- R01 AI136514/AI/NIAID NIH HHS/United States
- R01 HL087690/HL/NHLBI NIH HHS/United States
- P30 CA015704/CA/NCI NIH HHS/United States
- S10 OD028685/OD/NIH HHS/United States
- R01 AI107625/AI/NIAID NIH HHS/United States
- R01 AI120997/AI/NIAID NIH HHS/United States
- R01 AI146028/AI/NIAID NIH HHS/United States
- 75N93021C00016/AI/NIAID NIH HHS/United States
- R01 HL088201/HL/NHLBI NIH HHS/United States
- HHMI/Howard Hughes Medical Institute/United States
LinkOut - more resources
Full Text Sources
