

TFEB- and TFE3-dependent autophagy activation supports cancer proliferation in the absence of centrosomes
- PMID: 35316161
- PMCID: PMC9673955
- DOI: 10.1080/15548627.2022.2051880
TFEB- and TFE3-dependent autophagy activation supports cancer proliferation in the absence of centrosomes
Abstract
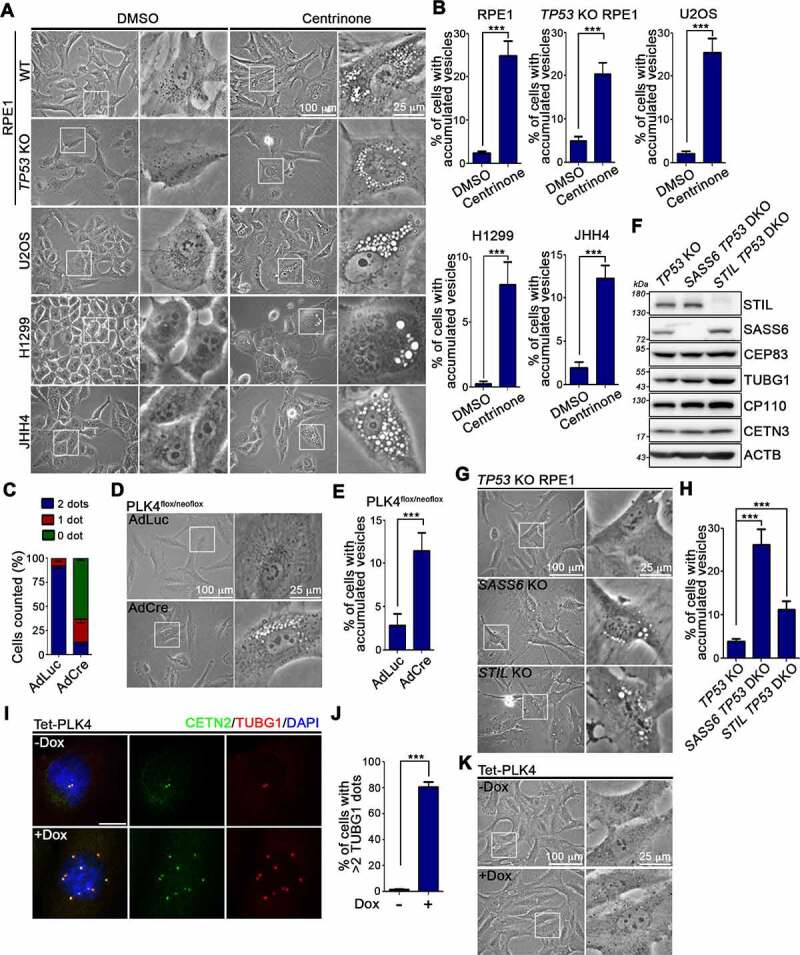
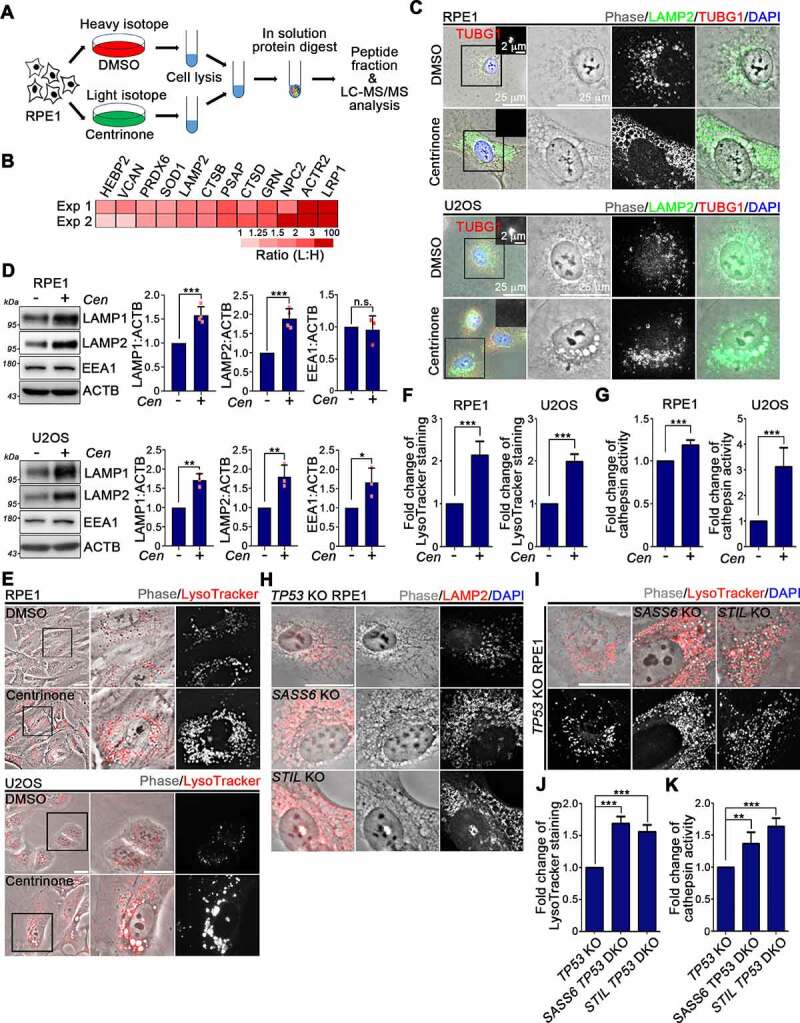
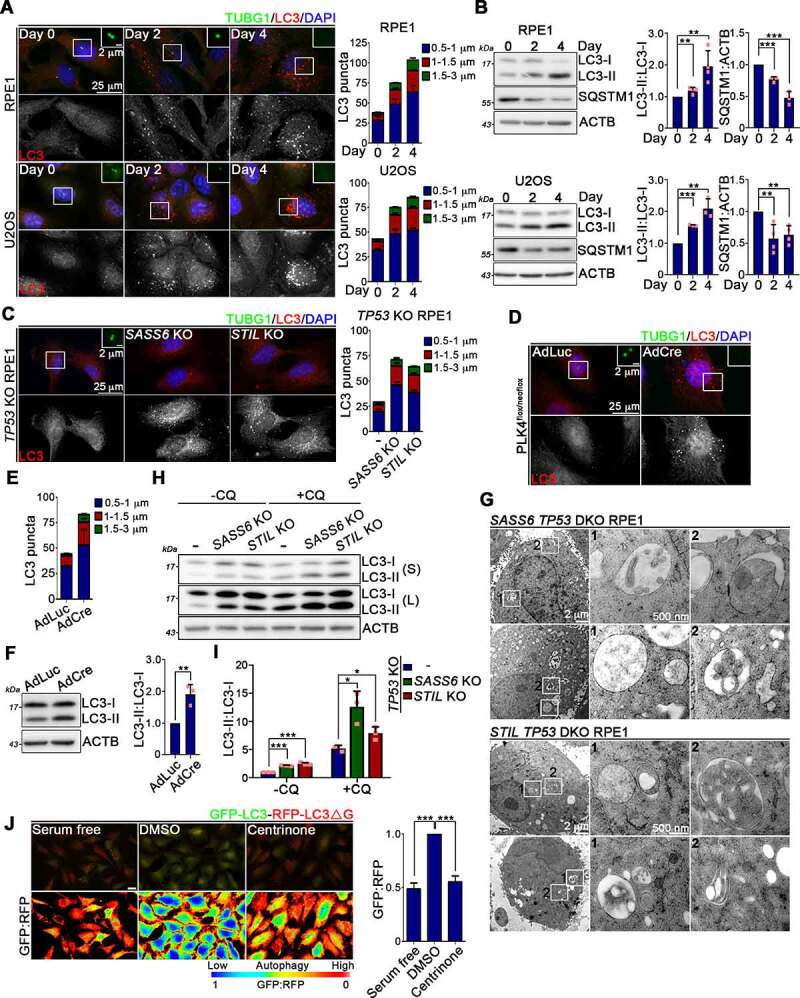
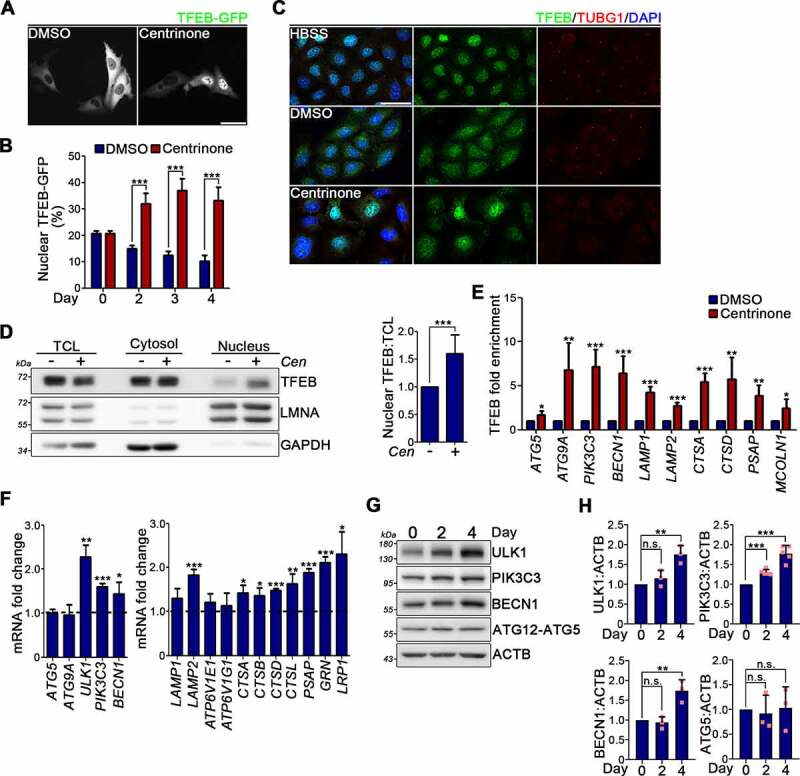
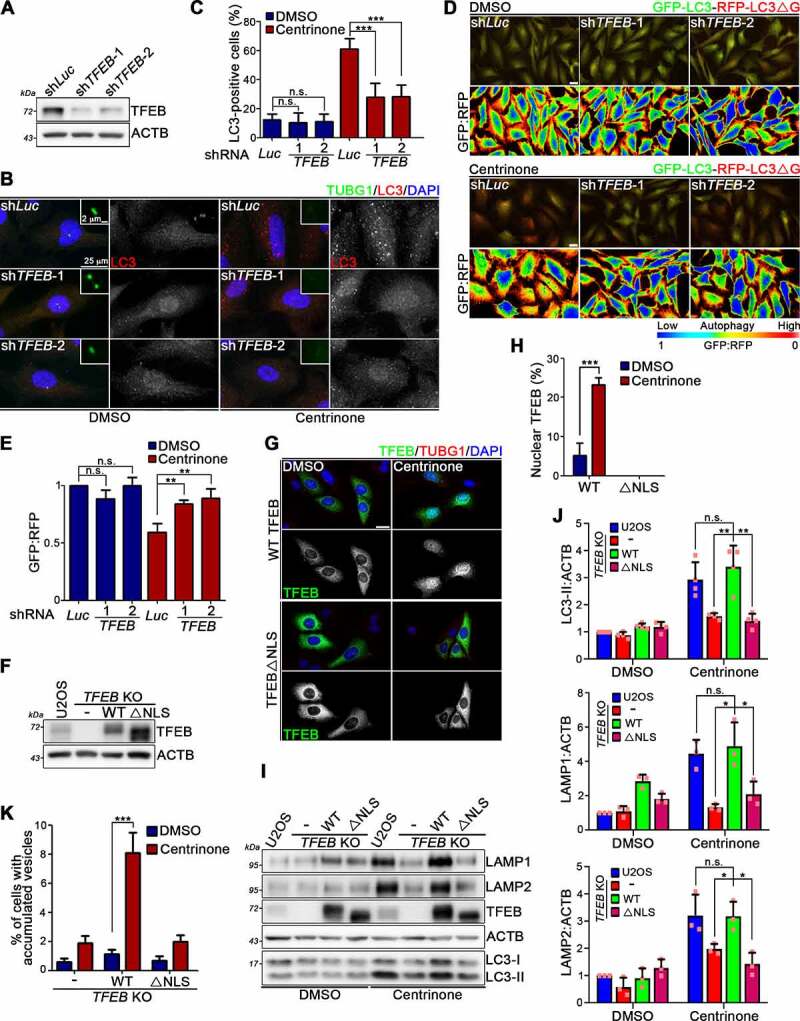
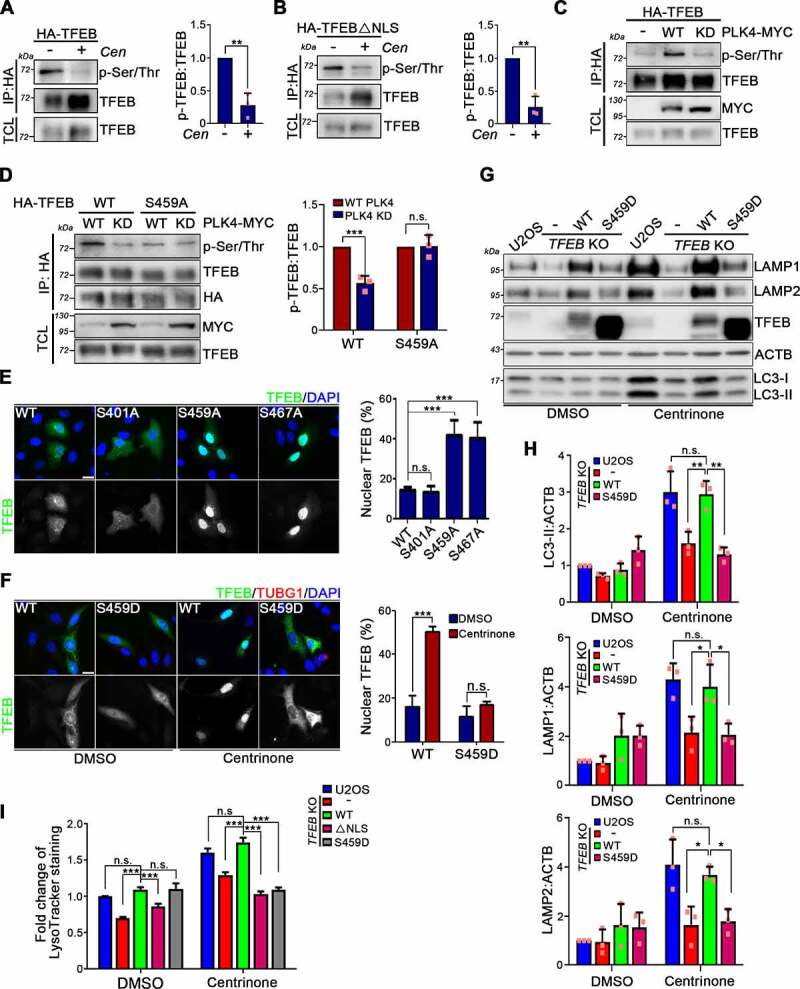
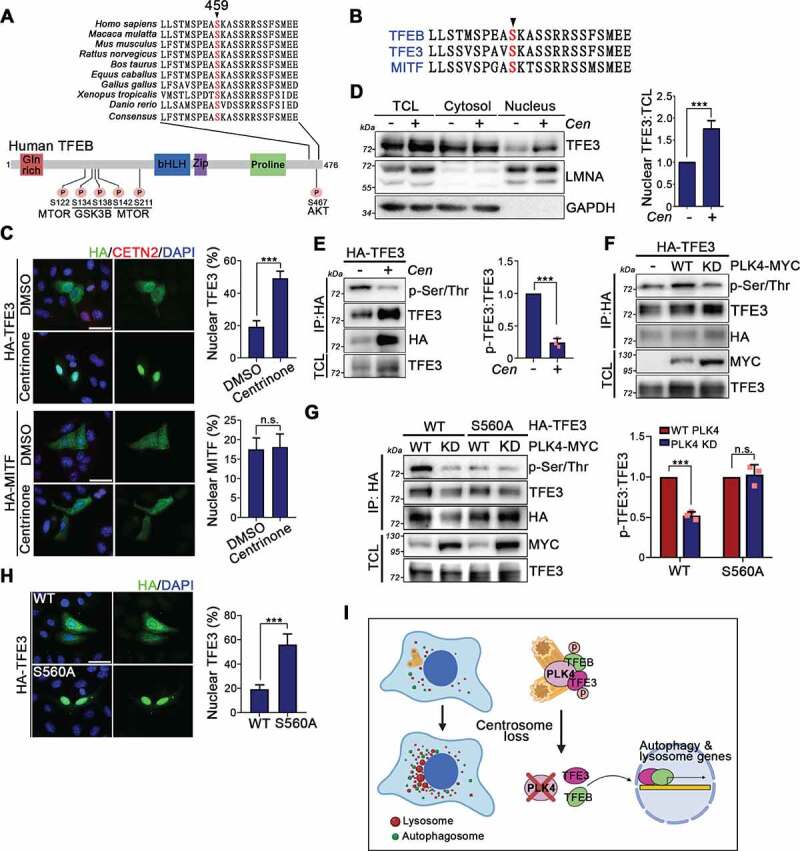
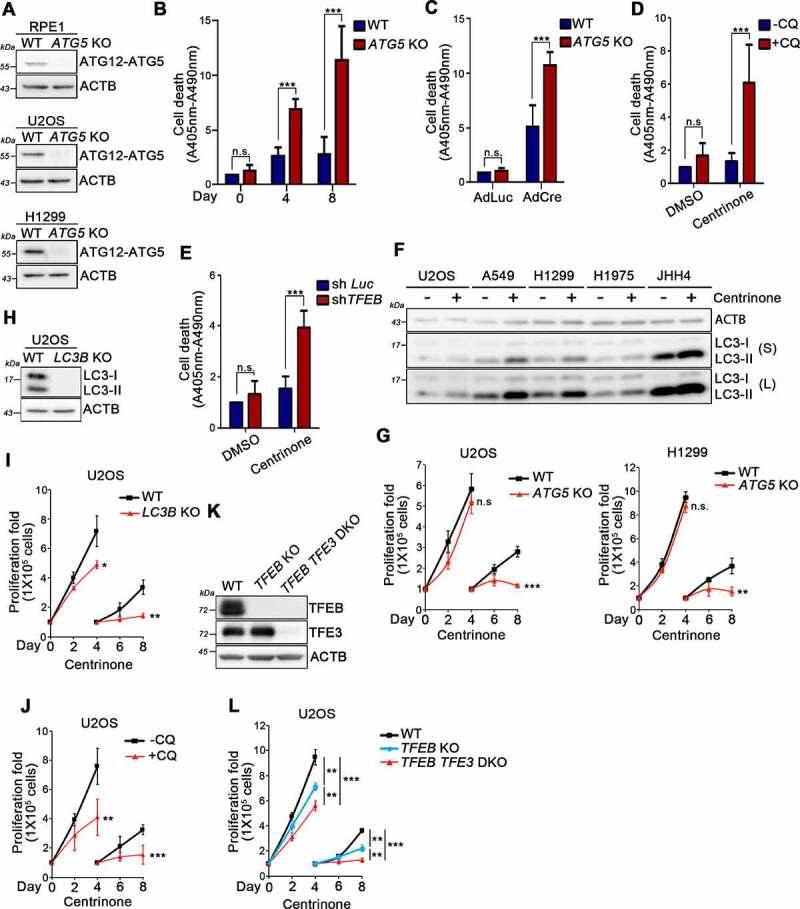
Centrosome amplification is a phenomenon frequently observed in human cancers, so centrosome depletion has been proposed as a therapeutic strategy. However, despite being afflicted with a lack of centrosomes, many cancer cells can still proliferate, implying there are impediments to adopting centrosome depletion as a treatment strategy. Here, we show that TFEB- and TFE3-dependent autophagy activation contributes to acentrosomal cancer proliferation. Our biochemical analyses uncover that both TFEB and TFE3 are novel PLK4 (polo like kinase 4) substrates. Centrosome depletion inactivates PLK4, resulting in TFEB and TFE3 dephosphorylation and subsequent promotion of TFEB and TFE3 nuclear translocation and transcriptional activation of autophagy- and lysosome-related genes. A combination of centrosome depletion and inhibition of the TFEB-TFE3 autophagy-lysosome pathway induced strongly anti-proliferative effects in cancer cells. Thus, our findings point to a new strategy for combating cancer.Abbreviations: AdCre: adenoviral Cre recombinase; AdLuc: adenoviral luciferase; ATG5: autophagy related 5; CQ: chloroquine; DAPI: 4',6-diamidino-2-phenylindole; DKO: double knockout; GFP: green fluorescent protein; KO: knockout; LAMP1: lysosomal associated membrane protein 1; LAMP2: lysosomal associated membrane protein 2; LTR: LysoTracker Red; MAP1LC3B/LC3B: microtubule associated protein 1 light chain 3 beta; MITF: melanocyte inducing transcription factor; PLK4: polo like kinase 4; RFP: red fluorescent protein; SASS6: SAS-6 centriolar assembly protein; STIL: STIL centriolar assembly protein; TFEB: transcription factor EB; TFEBΔNLS: TFEB lacking a nuclear localization signal; TFE3: transcription factor binding to IGHM enhancer 3; TP53/p53: tumor protein p53.
Keywords: Anti-cancer therapy; PLK4; autophagy; centrosome; lysosomal biogenesis; transcription factor E3; transcription factor EB.
Conflict of interest statement
No potential conflict of interest was reported by the author(s).
Figures
Similar articles
-
AMPK-dependent phosphorylation is required for transcriptional activation of TFEB and TFE3.Autophagy. 2021 Dec;17(12):3957-3975. doi: 10.1080/15548627.2021.1898748. Epub 2021 Mar 18. Autophagy. 2021. PMID: 33734022 Free PMC article.
-
Phosphorylation of EIF2S1 (eukaryotic translation initiation factor 2 subunit alpha) is indispensable for nuclear translocation of TFEB and TFE3 during ER stress.Autophagy. 2023 Jul;19(7):2111-2142. doi: 10.1080/15548627.2023.2173900. Epub 2023 Feb 9. Autophagy. 2023. PMID: 36719671 Free PMC article.
-
Impaired TFEB-mediated lysosomal biogenesis promotes the development of pancreatitis in mice and is associated with human pancreatitis.Autophagy. 2019 Nov;15(11):1954-1969. doi: 10.1080/15548627.2019.1596486. Epub 2019 Mar 30. Autophagy. 2019. PMID: 30894069 Free PMC article.
-
TFEB at a glance.J Cell Sci. 2016 Jul 1;129(13):2475-81. doi: 10.1242/jcs.146365. Epub 2016 Jun 1. J Cell Sci. 2016. PMID: 27252382 Free PMC article. Review.
-
TFEB Biology and Agonists at a Glance.Cells. 2021 Feb 5;10(2):333. doi: 10.3390/cells10020333. Cells. 2021. PMID: 33562649 Free PMC article. Review.
Cited by
-
Autophagy: a critical mechanism of N6-methyladenosine modification involved in tumor progression and therapy resistance.Cell Death Dis. 2024 Oct 28;15(10):783. doi: 10.1038/s41419-024-07148-w. Cell Death Dis. 2024. PMID: 39468015 Free PMC article. Review.
-
TRIM27 elicits protective immunity against tuberculosis by activating TFEB-mediated autophagy flux.Autophagy. 2024 Jul;20(7):1483-1504. doi: 10.1080/15548627.2024.2321831. Epub 2024 Mar 4. Autophagy. 2024. PMID: 38390831 Free PMC article.
-
Corynoxine promotes TFEB/TFE3-mediated autophagy and alleviates Aβ pathology in Alzheimer's disease models.Acta Pharmacol Sin. 2024 May;45(5):900-913. doi: 10.1038/s41401-023-01197-1. Epub 2024 Jan 15. Acta Pharmacol Sin. 2024. PMID: 38225393 Free PMC article.
-
STIL overexpression shortens lifespan and reduces tumor formation in mice.PLoS Genet. 2024 Oct 28;20(10):e1011460. doi: 10.1371/journal.pgen.1011460. eCollection 2024 Oct. PLoS Genet. 2024. PMID: 39466849 Free PMC article.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous