

Validation-Based Insertional Mutagenesis (VBIM), A Powerful Forward Genetic Screening Strategy
- PMID: 35316583
- PMCID: PMC8969887
- DOI: 10.1002/cpz1.394
Validation-Based Insertional Mutagenesis (VBIM), A Powerful Forward Genetic Screening Strategy
Abstract
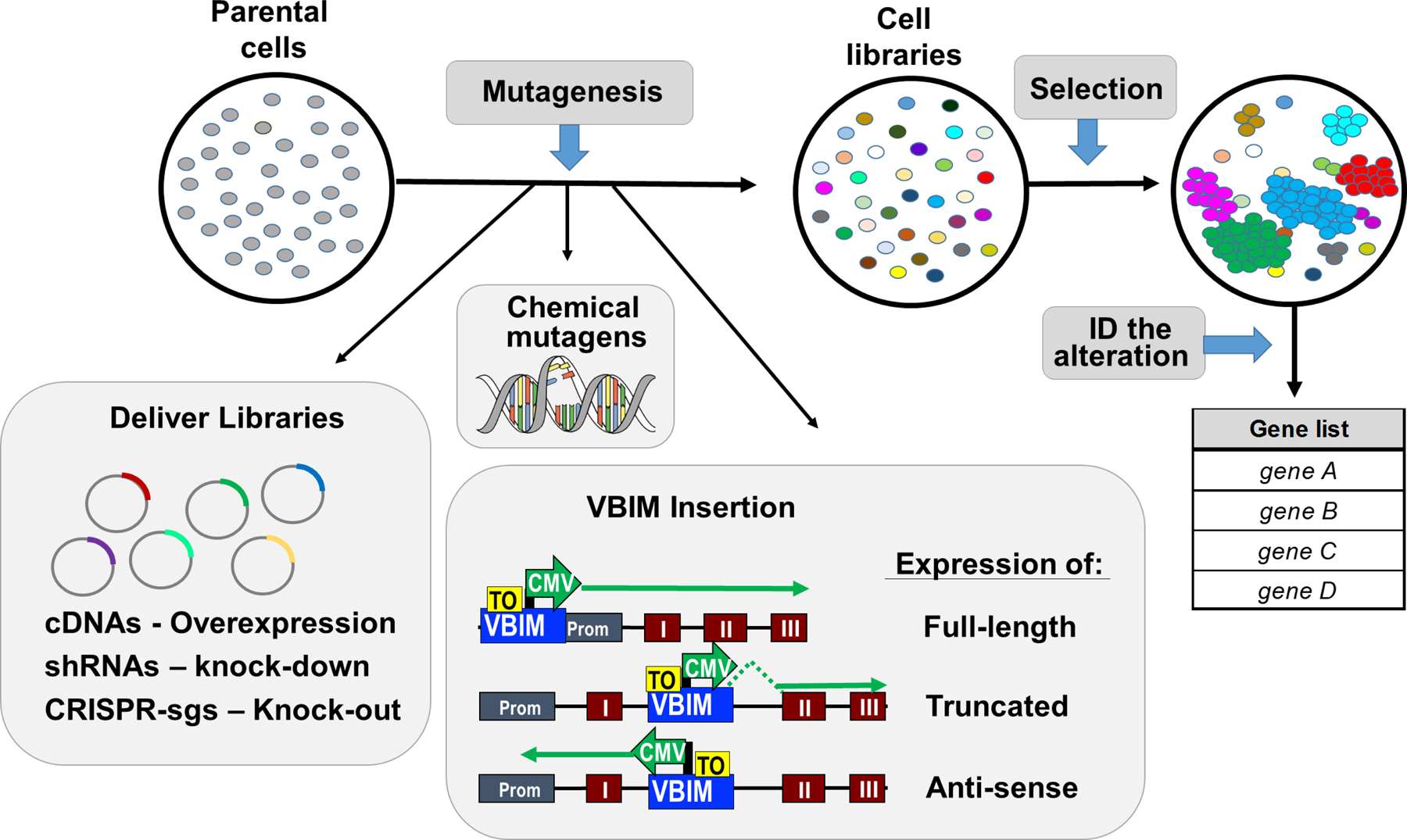
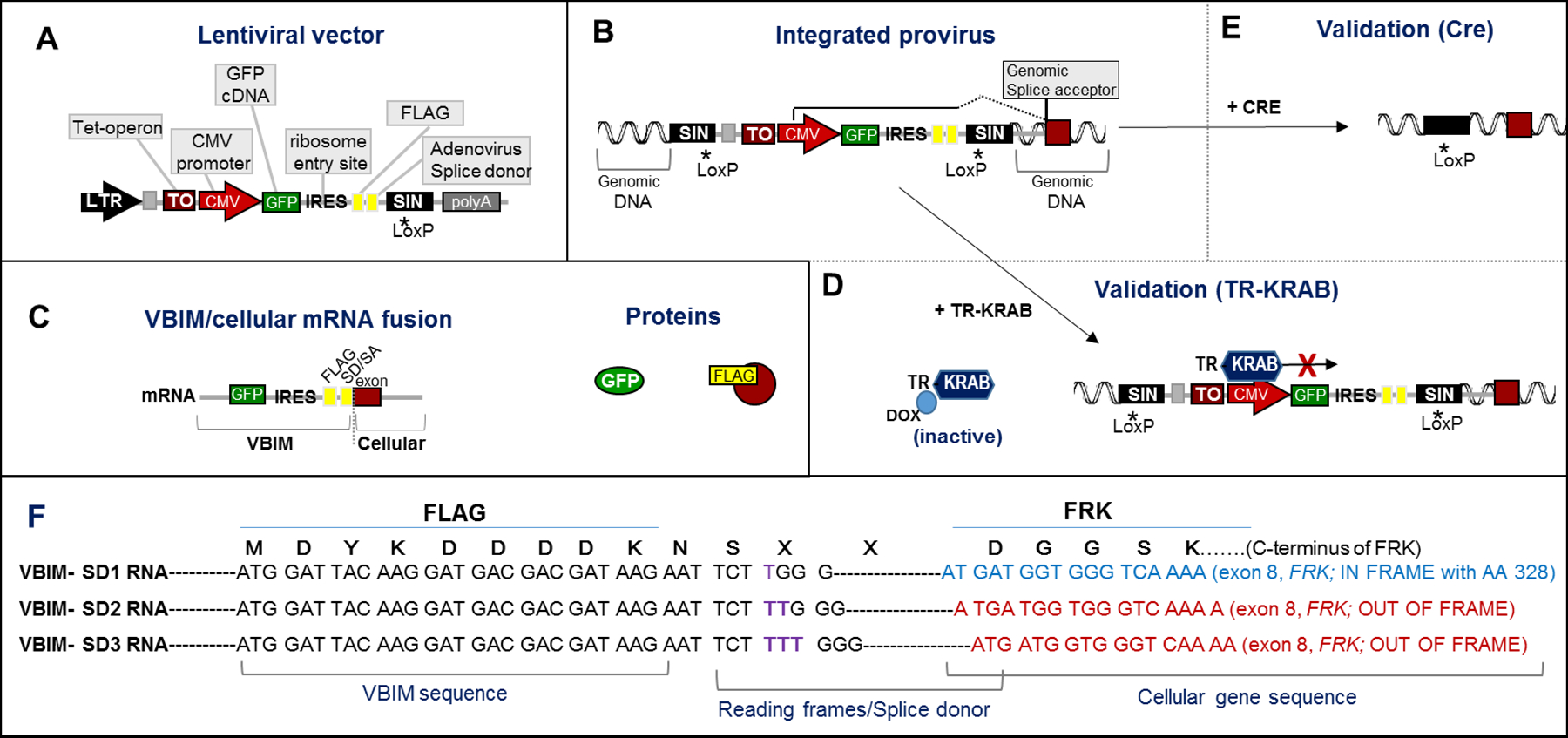
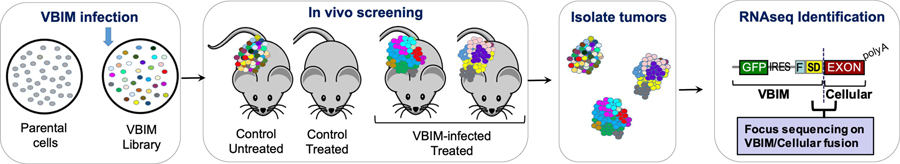
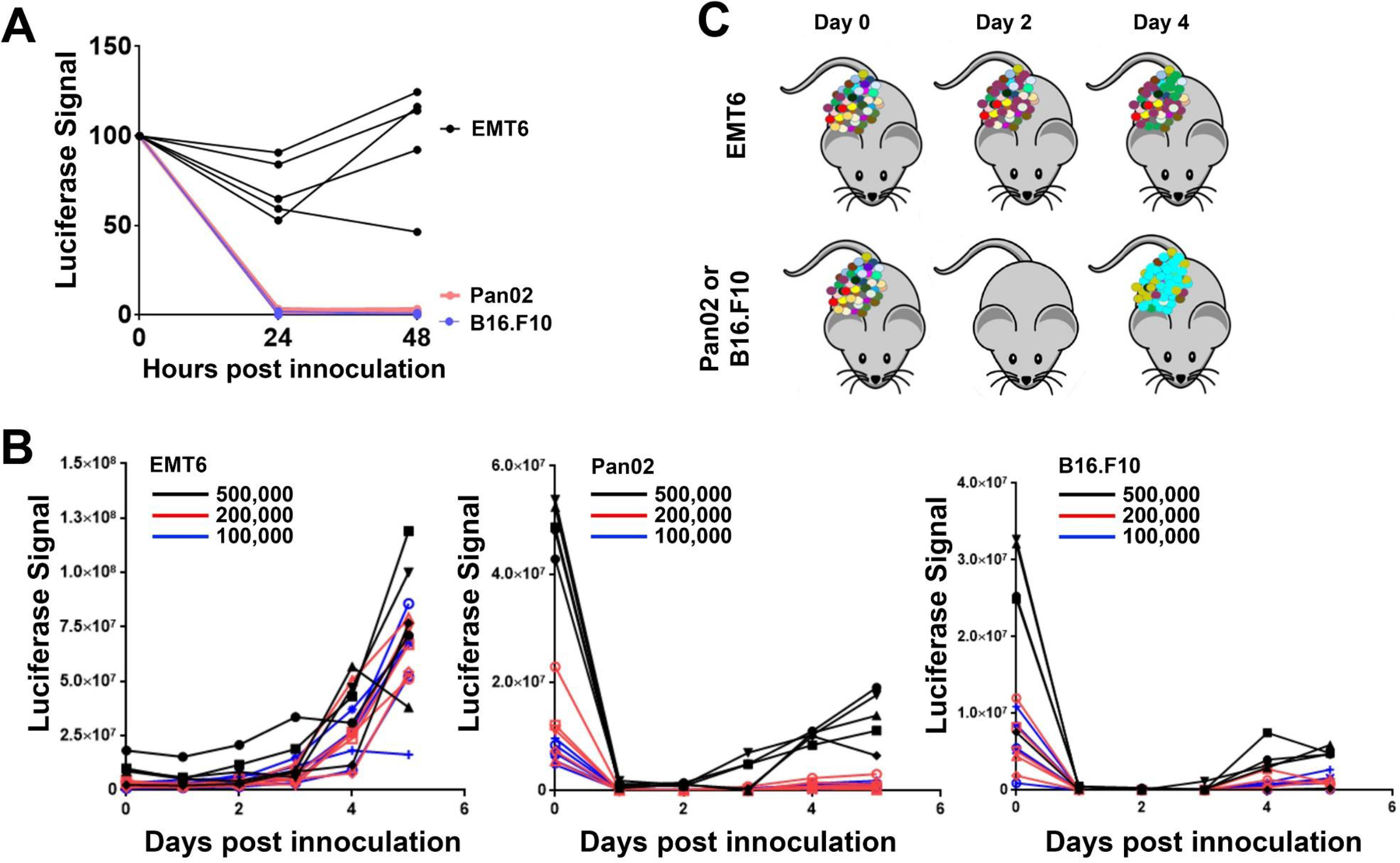
Forward genetics begins with a biological phenotype and attempts to identify genetic changes that influence that phenotype. These changes can be induced in a selected group of genes, for instance, by using libraries of cDNAs, shRNAs, CRISPR guide RNAs, or genetic suppressor elements (GSEs), or randomly throughout the genome using chemical or insertional mutagens, with each approach creating distinct genetic changes. The Validation-Based Insertional Mutagenesis (VBIM) strategy utilizes modified lentiviruses as insertional mutagens, placing strong promoters throughout the genome. Generating libraries with millions of cells carrying one or a few VBIM promoter insertions is straightforward, allowing selection of cells in which overexpression of VBIM-driven RNAs or proteins promote the phenotype of interest. VBIM-driven RNAs may encode full-length proteins, truncated proteins (which may have wild-type, constitutive, or dominant-negative activity), or antisense RNAs that can disrupt gene expression. The diversity in VBIM-driven changes allows for the identification of both gain-of-function and loss-of-function mutations in a single screen. Additionally, VBIM can target any genomic locus, regardless of whether it is expressed in the cells under study or known to have a biological function, allowing for true whole-genome screens without the complication and cost of constructing, maintaining, and delivering a comprehensive library. Here, we review the VBIM strategy and discuss examples in which VBIM has been successfully used in diverse screens to identify novel genes or novel functions for known genes. In addition, we discuss considerations for transitioning the VBIM strategy to in vivo screens. We hope that other laboratories will be encouraged to use the VBIM strategy to identify genes that influence their phenotypes of interest. © 2022 Wiley Periodicals LLC.
Keywords: cell libraries; drug resistance; forward genetics; in vivo screens; insertional mutagenesis; lentiviral vectors.
© 2022 Wiley Periodicals LLC.
Conflict of interest statement
CONFLICT OF INTEREST STATEMENT
The authors declare no conflicts of interest.
Figures

Similar articles
-
Validation-based insertional mutagenesis for identification of Nup214 as a host factor for EV71 replication in RD cells.Biochem Biophys Res Commun. 2013 Aug 2;437(3):452-6. doi: 10.1016/j.bbrc.2013.06.101. Epub 2013 Jul 3. Biochem Biophys Res Commun. 2013. PMID: 23831628
-
Forward genetic screening for regulators involved in cholesterol synthesis using validation-based insertional mutagenesis.PLoS One. 2014 Nov 26;9(11):e112632. doi: 10.1371/journal.pone.0112632. eCollection 2014. PLoS One. 2014. PMID: 25426949 Free PMC article.
-
Validation-based insertional mutagenesis identifies lysine demethylase FBXL11 as a negative regulator of NFkappaB.Proc Natl Acad Sci U S A. 2009 Sep 22;106(38):16339-44. doi: 10.1073/pnas.0908560106. Epub 2009 Sep 1. Proc Natl Acad Sci U S A. 2009. PMID: 19805303 Free PMC article.
-
Cancer gene discovery: exploiting insertional mutagenesis.Mol Cancer Res. 2013 Oct;11(10):1141-58. doi: 10.1158/1541-7786.MCR-13-0244. Epub 2013 Aug 8. Mol Cancer Res. 2013. PMID: 23928056 Free PMC article. Review.
-
Forward and Reverse Genetics of B Cell Malignancies: From Insertional Mutagenesis to CRISPR-Cas.Front Immunol. 2021 Aug 13;12:670280. doi: 10.3389/fimmu.2021.670280. eCollection 2021. Front Immunol. 2021. PMID: 34484175 Free PMC article. Review.
References
-
- Cipriano R, Miskimen KLS, Bryson BL, Foy CR, Bartel CA, & Jackson MW (2013). FAM83B-mediated activation of PI3K/AKT and MAPK signaling cooperates to promote epithelial cell transformation and resistance to targeted therapies. Oncotarget, 4(5), 729–738. Retrieved from http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3742833/ - PMC - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
