

The glucose transporter GLUT3 controls T helper 17 cell responses through glycolytic-epigenetic reprogramming
- PMID: 35316657
- PMCID: PMC9019065
- DOI: 10.1016/j.cmet.2022.02.015
The glucose transporter GLUT3 controls T helper 17 cell responses through glycolytic-epigenetic reprogramming
Abstract
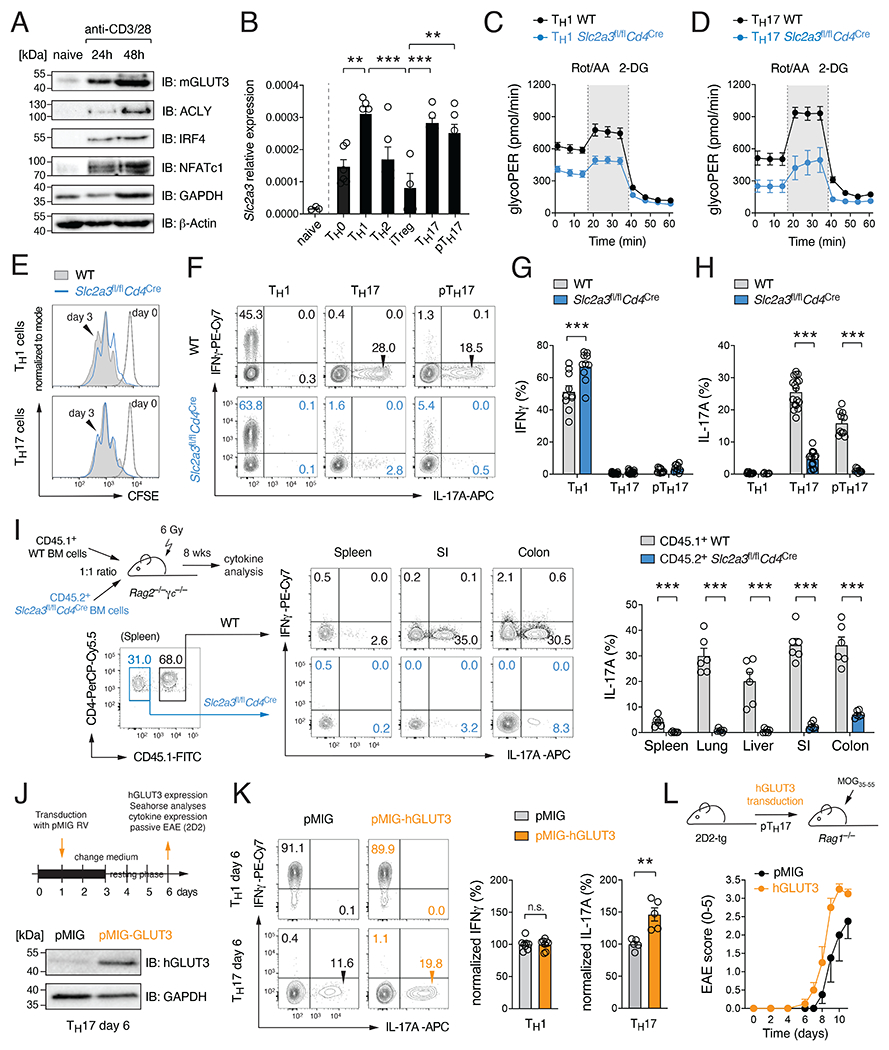
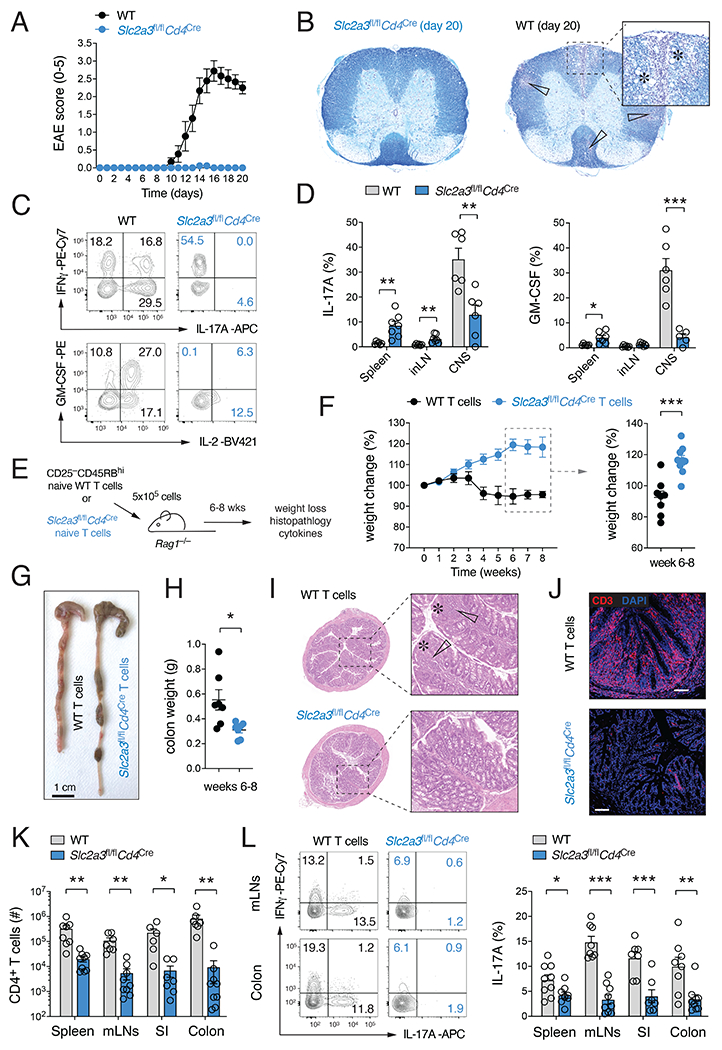
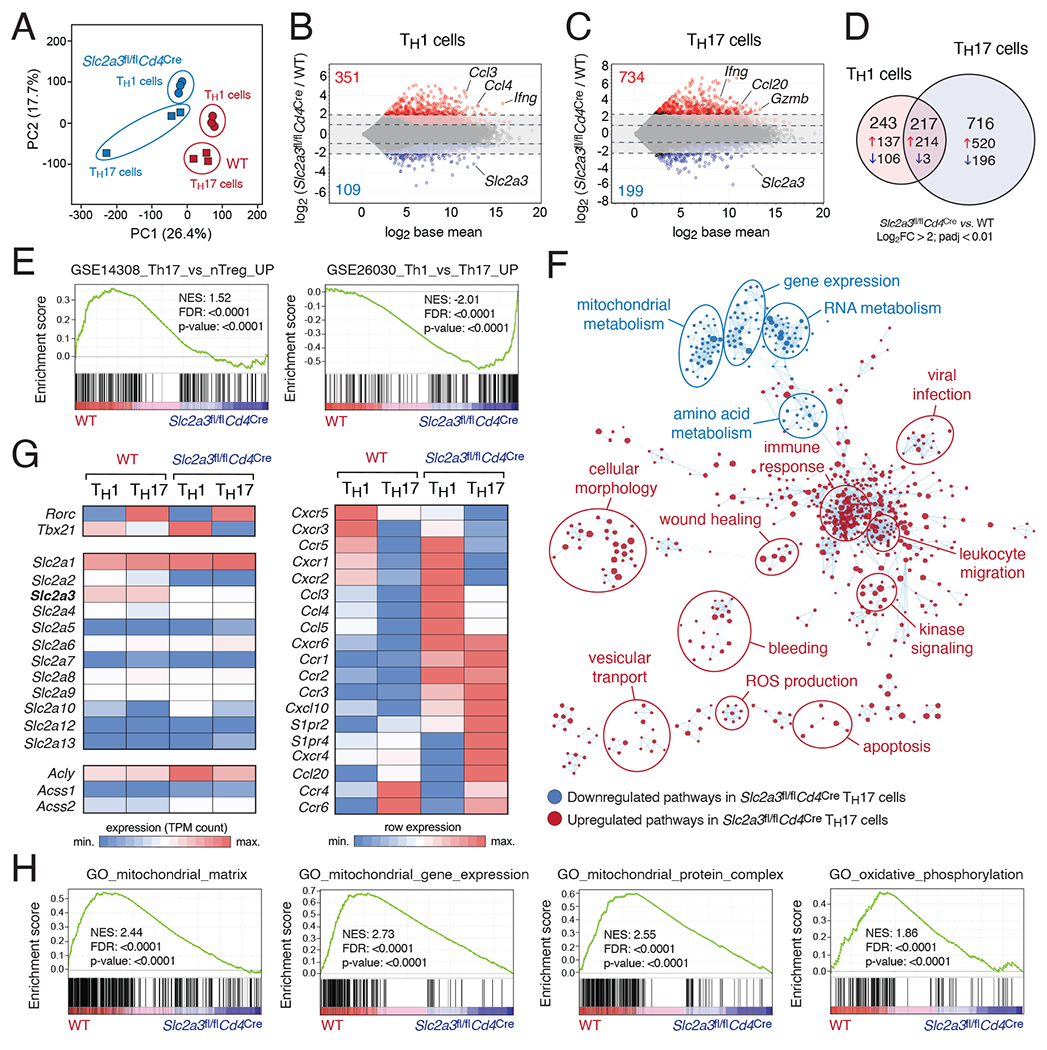
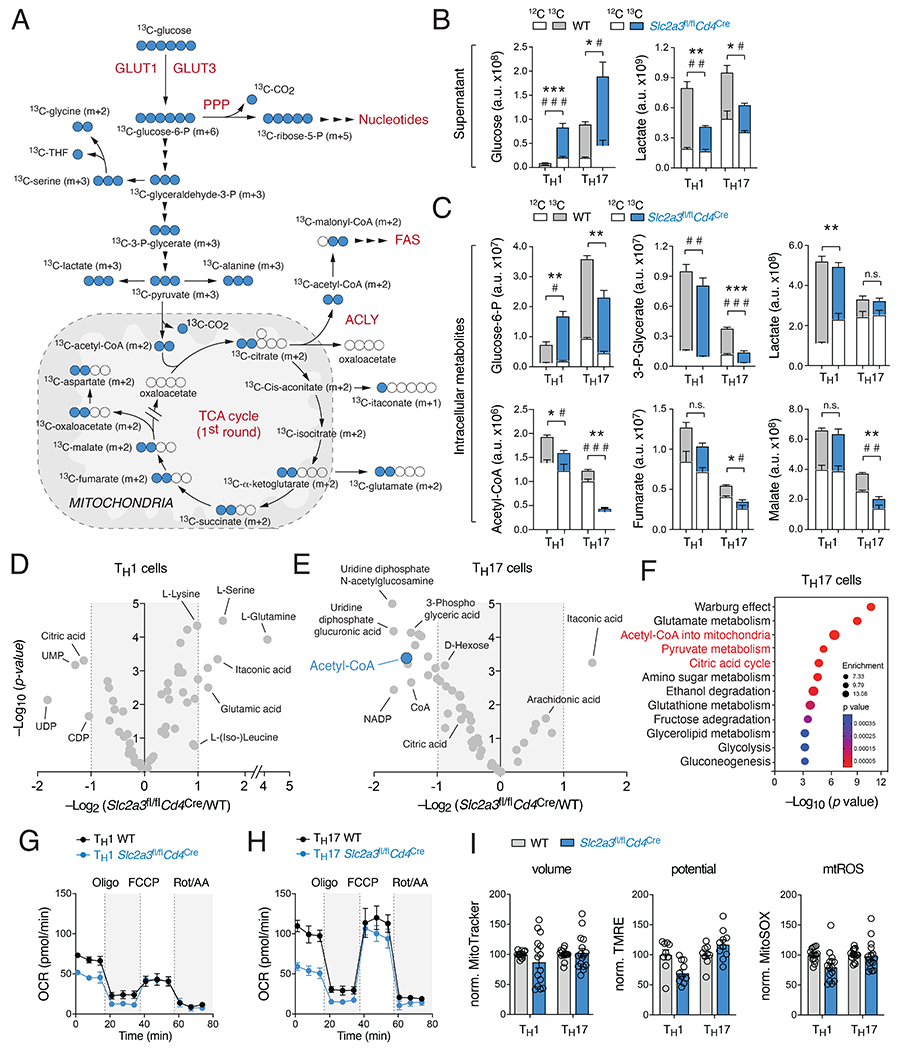
Metabolic reprogramming is a hallmark of activated T cells. The switch from oxidative phosphorylation to aerobic glycolysis provides energy and intermediary metabolites for the biosynthesis of macromolecules to support clonal expansion and effector function. Here, we show that glycolytic reprogramming additionally controls inflammatory gene expression via epigenetic remodeling. We found that the glucose transporter GLUT3 is essential for the effector functions of Th17 cells in models of autoimmune colitis and encephalomyelitis. At the molecular level, we show that GLUT3-dependent glucose uptake controls a metabolic-transcriptional circuit that regulates the pathogenicity of Th17 cells. Metabolomic, epigenetic, and transcriptomic analyses linked GLUT3 to mitochondrial glucose oxidation and ACLY-dependent acetyl-CoA generation as a rate-limiting step in the epigenetic regulation of inflammatory gene expression. Our findings are also important from a translational perspective because inhibiting GLUT3-dependent acetyl-CoA generation is a promising metabolic checkpoint to mitigate Th17-cell-mediated inflammatory diseases.
Keywords: ACLY; ATP-citrate lyase; GLUT1; GLUT3; Th17 cells; acetyl-CoA; glucose metabolism; glycolysis; histone acetylation; immunometabolism.
Copyright © 2022 Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests The authors declare no competing interests.
Figures
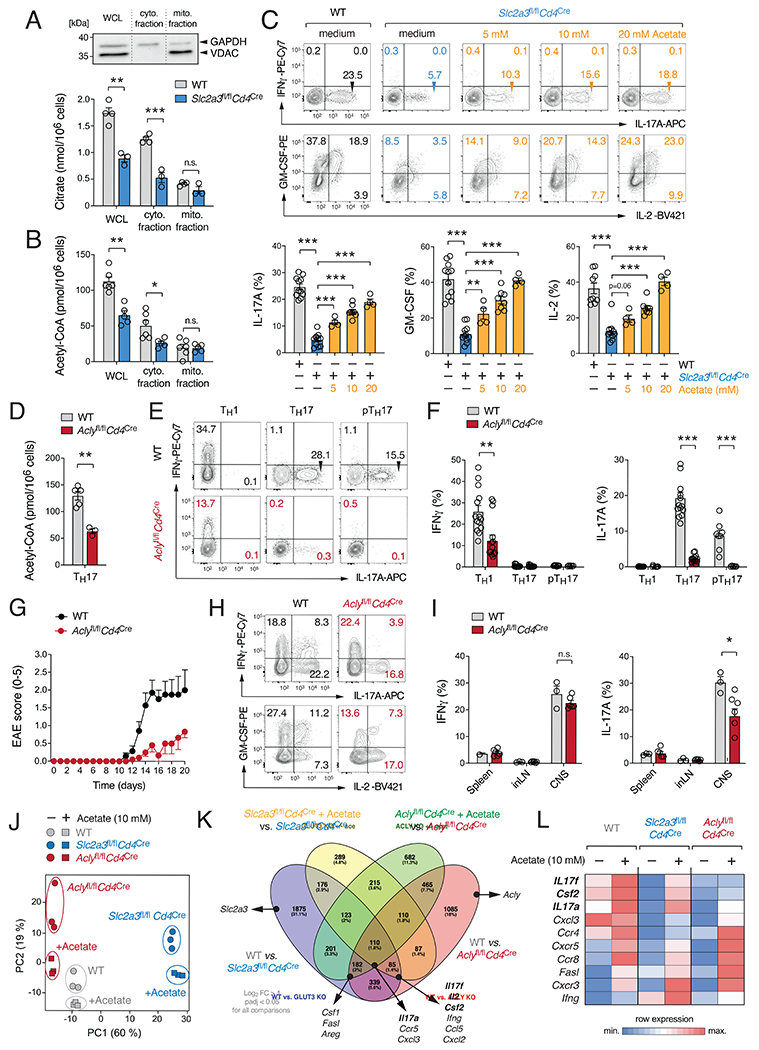
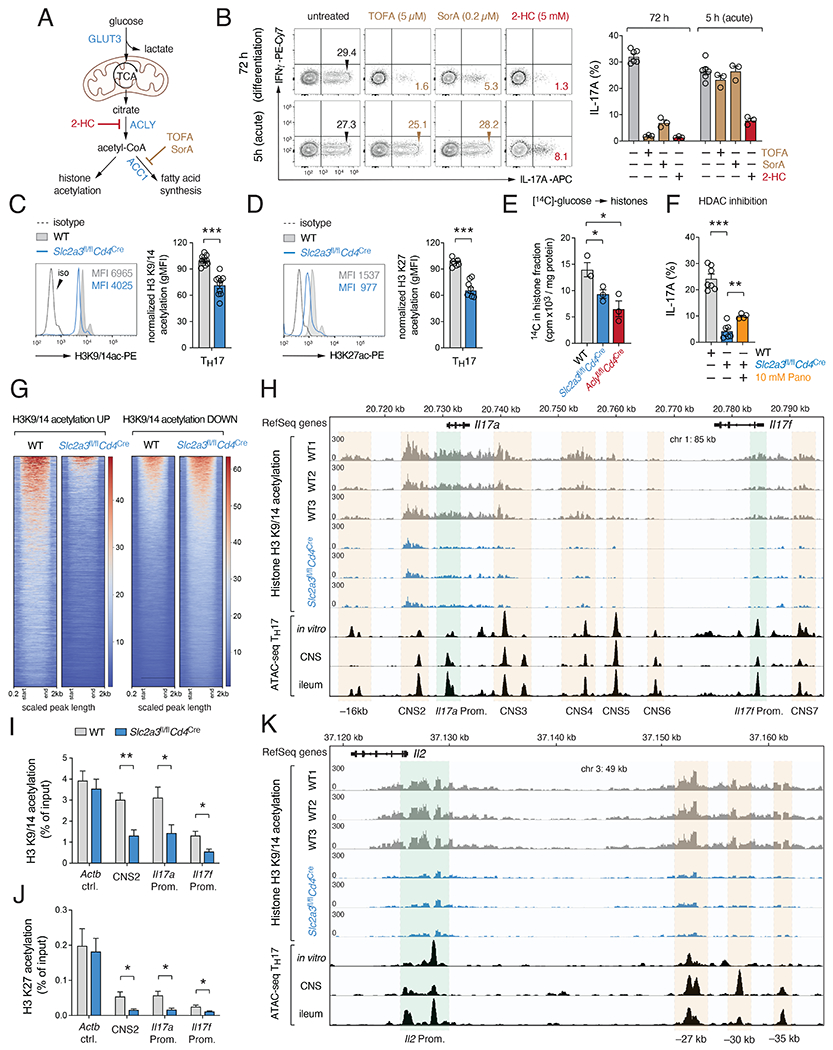
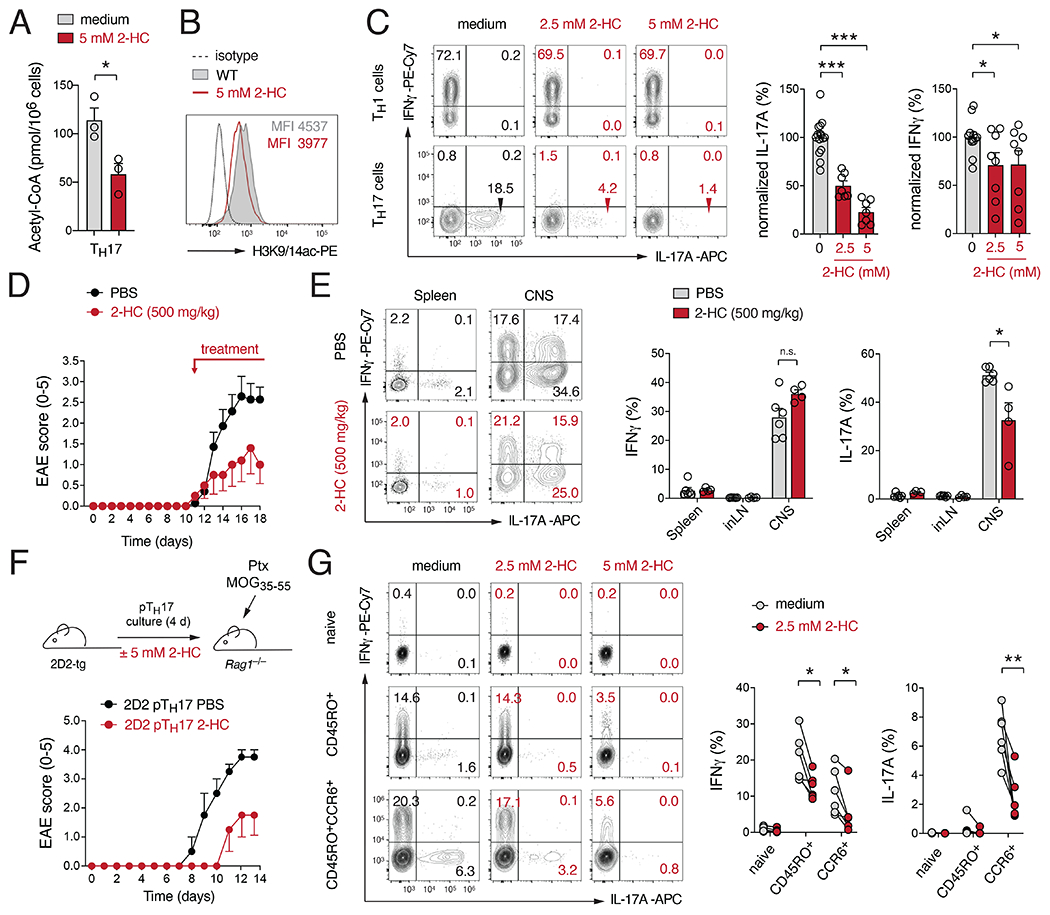
Comment in
-
Sugar addiction: An Achilles' heel of auto-immune diseases?Cell Metab. 2022 Apr 5;34(4):503-505. doi: 10.1016/j.cmet.2022.03.007. Cell Metab. 2022. PMID: 35385700
References
-
- Akimzhanov AM, Yang XO, and Dong C (2007). Chromatin remodeling of interleukin-17 (IL-17)-IL-17F cytokine gene locus during inflammatory helper T cell differentiation. J Biol Chem 282, 5969–5972. - PubMed
-
- Berod L, Friedrich C, Nandan A, Freitag J, Hagemann S, Harmrolfs K, Sandouk A, Hesse C, Castro CN, Bahre H, et al. (2014). De novo fatty acid synthesis controls the fate between regulatory T and T helper 17 cells. Nat Med 20, 1327–1333. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
