

A novel PD-L1-targeted shark VNAR single-domain-based CAR-T cell strategy for treating breast cancer and liver cancer
- PMID: 35317524
- PMCID: PMC8917269
- DOI: 10.1016/j.omto.2022.02.015
A novel PD-L1-targeted shark VNAR single-domain-based CAR-T cell strategy for treating breast cancer and liver cancer
Abstract
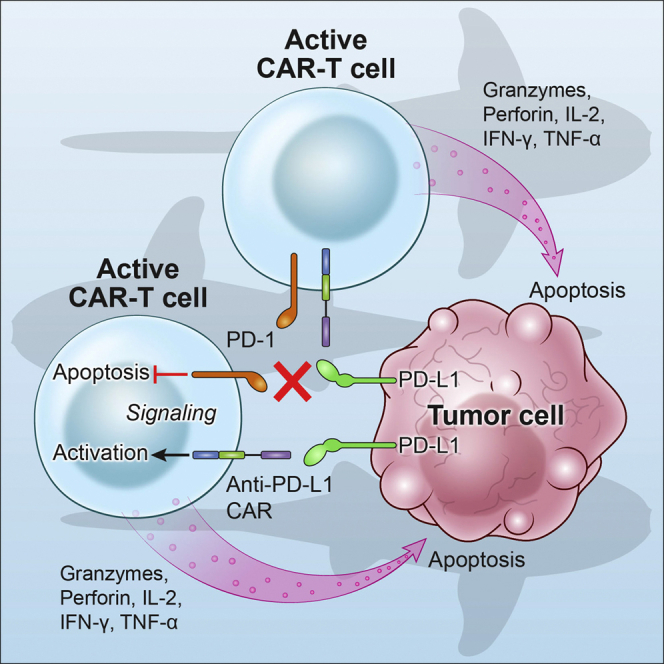
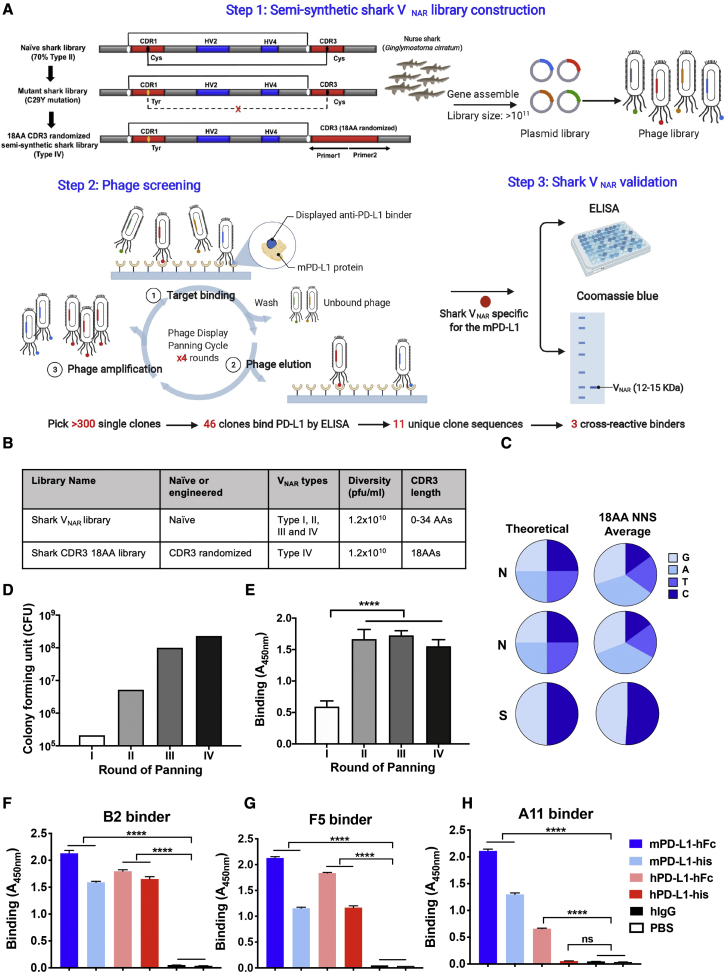
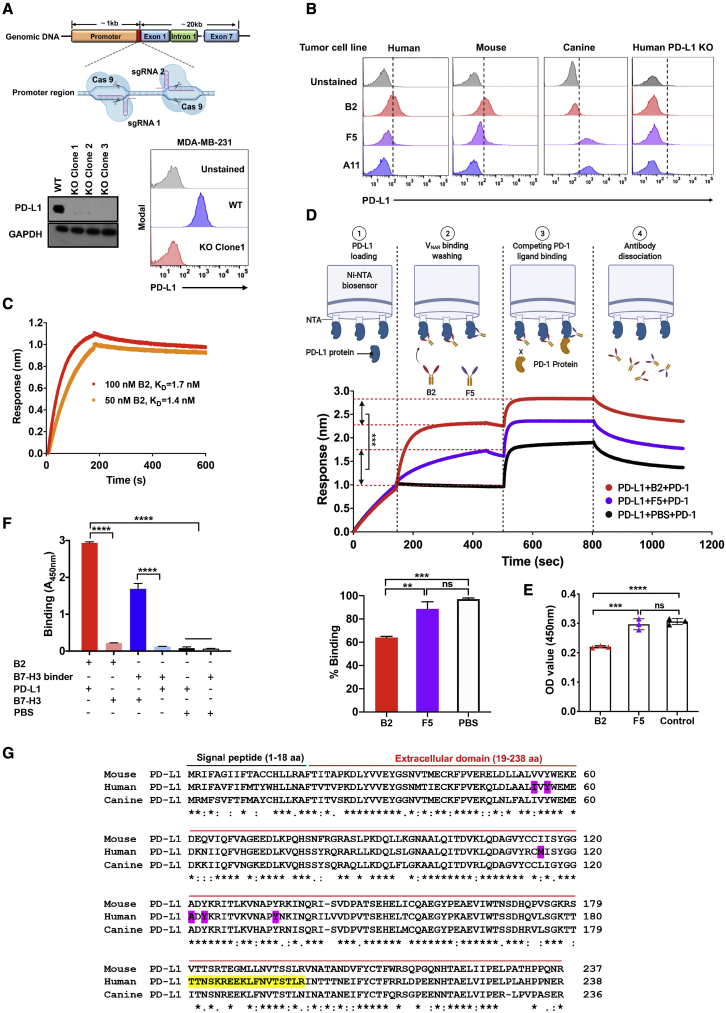
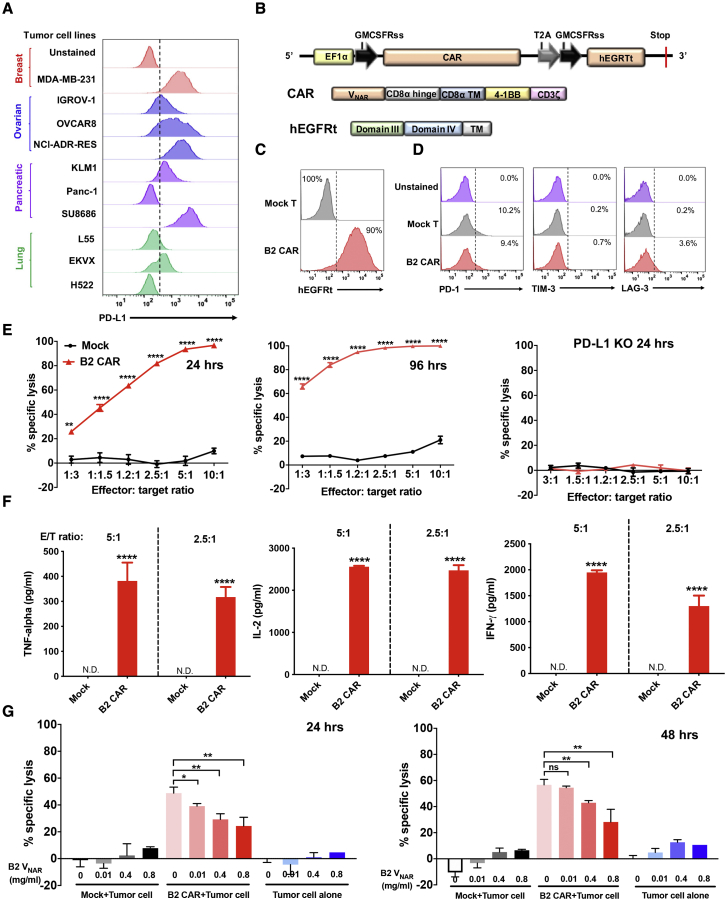
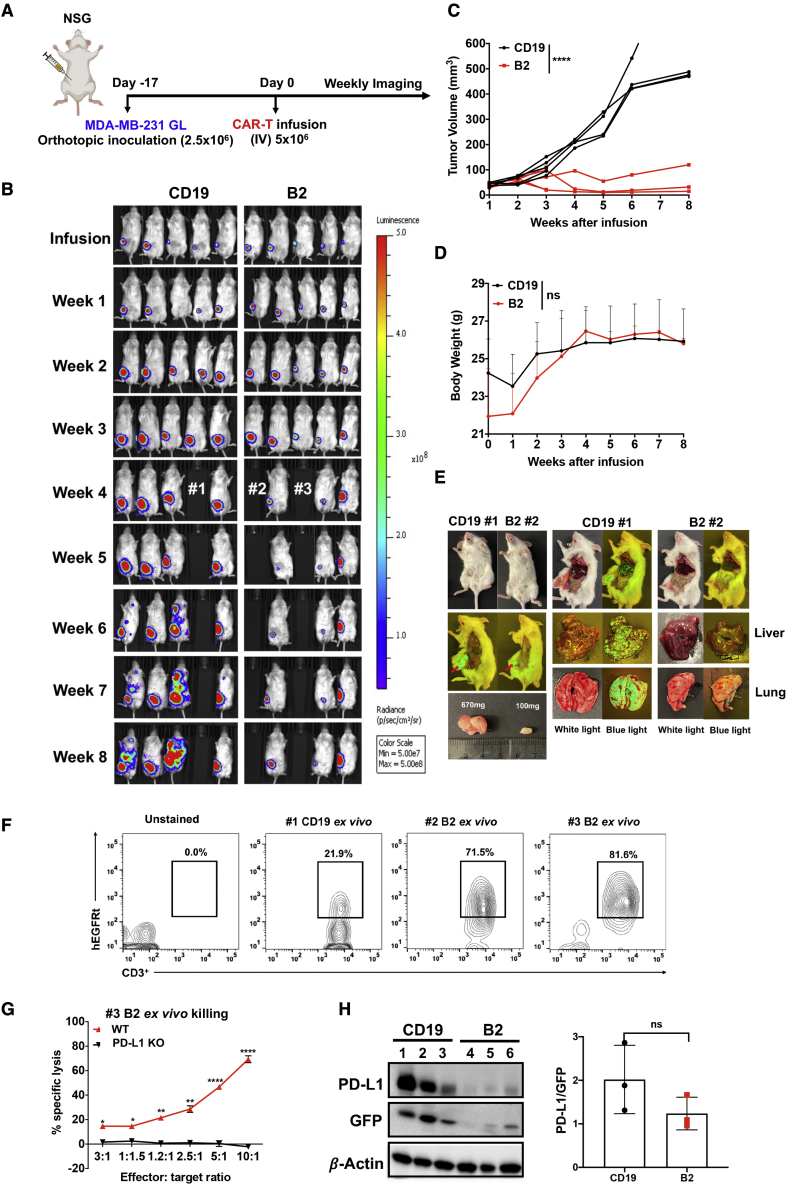
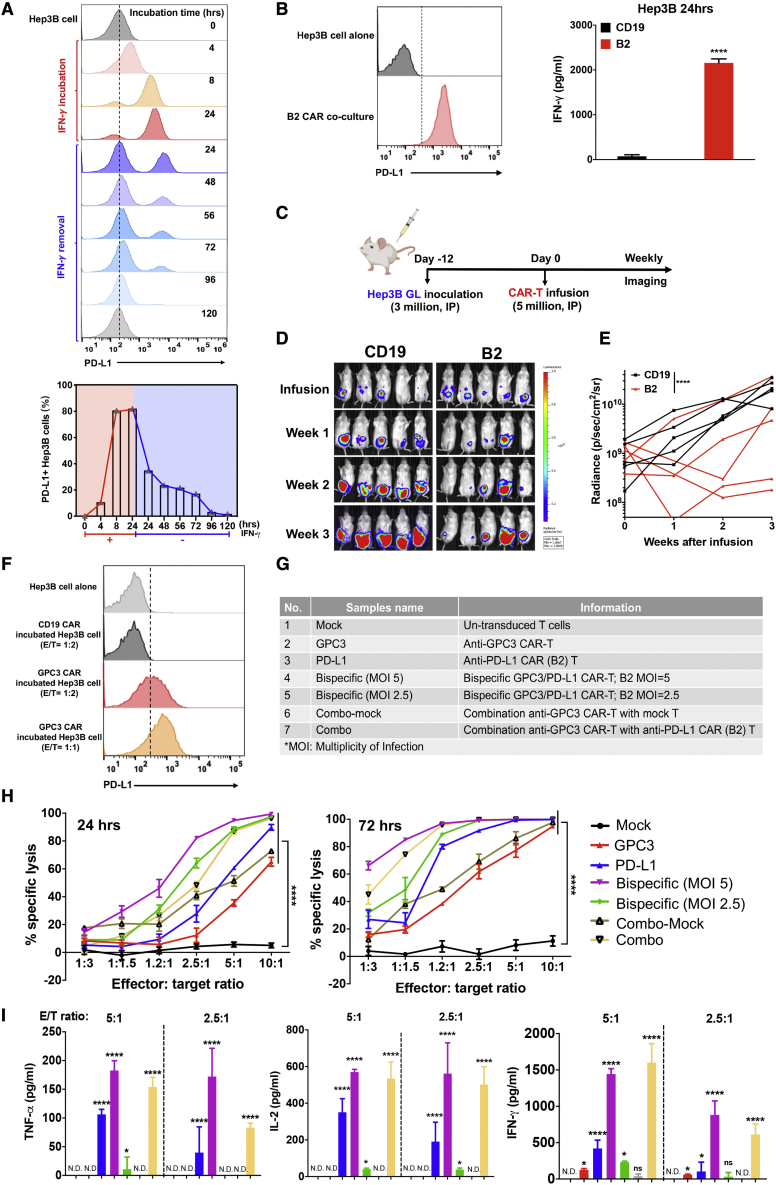
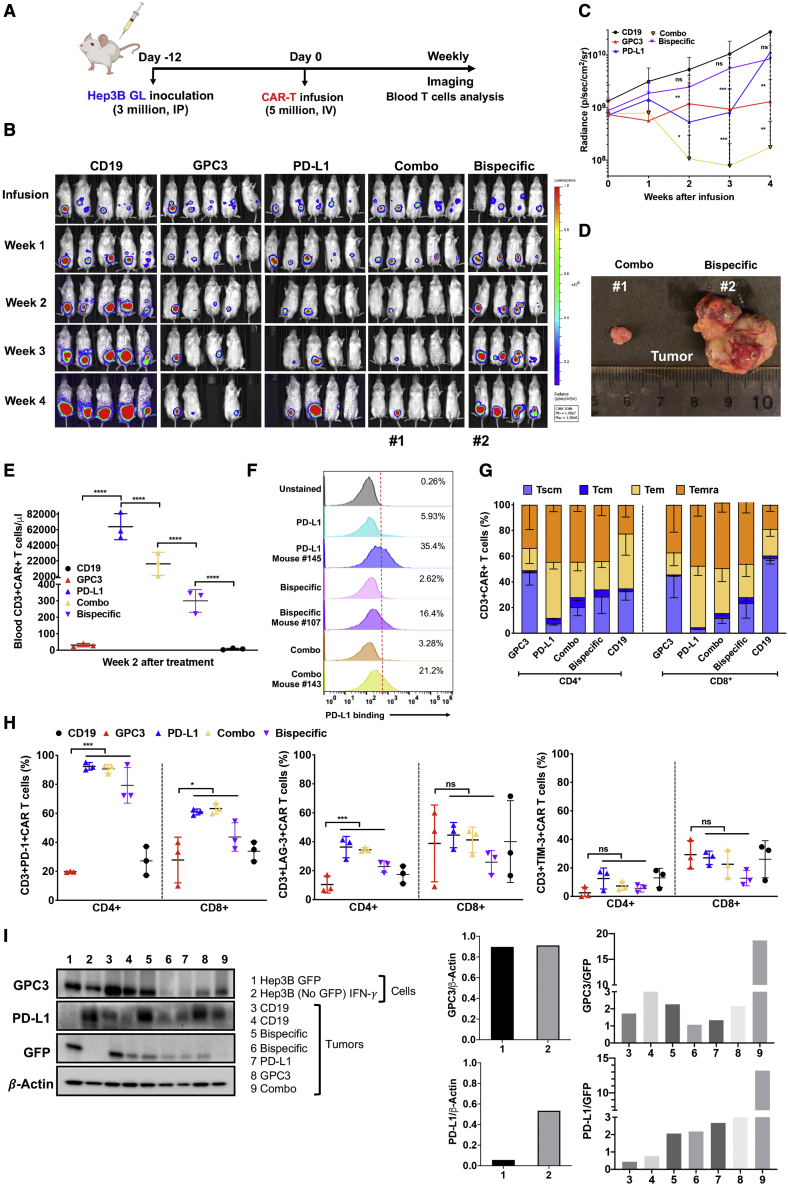
Chimeric antigen receptor (CAR)-T cell therapy shows excellent potency against hematological malignancies, but it remains challenging to treat solid tumors, mainly because of a lack of appropriate antigenic targets and an immunosuppressive tumor microenvironment (TME). The checkpoint molecule programmed death-ligand 1 (PD-L1) is widely overexpressed in multiple tumor types, and the programmed death-ligand 1 (PD-1)/PD-L1 interaction is a crucial mediator of immunosuppression in the TME. Here we constructed a semi-synthetic shark VNAR phage library and isolated anti-PD-L1 single-domain antibodies. Among these VNARs, B2 showed cross-reactivity to human, mouse, and canine PD-L1, and it partially blocked the interaction of human PD-1 with PD-L1. CAR (B2) T cells specifically lysed human breast cancer and liver cancer cells by targeting constitutive and inducible expression of PD-L1 and hindered tumor metastasis. Combination of PD-L1 CAR (B2) T cells with CAR T cells targeted by GPC3 (a liver cancer-specific antigen) regresses liver tumors in mice. We concluded that PD-L1-targeted shark VNAR single-domain-based CAR-T therapy is a novel strategy to treat breast and liver cancer. This study provides a rationale for potential use of PD-L1 CAR-T cells as a monotherapy or in combination with a tumor-specific therapy in clinical studies.
Keywords: CAR-T cells; GPC3; PD-L1; glypican-3; hepatocellular carcinoma; immune checkpoint; liver cancer; shark VNAR; single-domain antibody; triple-negative breast cancer.
Conflict of interest statement
M.H., G.M., D.L., H.E., and C.-P.D. are inventors on US provisional patent application no. 63/208,755 assigned to the NIH “Cross Species Single Domain Antibodies Targeting PD-L1 For Treating Solid Tumors.”
Figures
References
-
- Kochenderfer J.N., Wilson W.H., Janik J.E., Dudley M.E., Stetler-Stevenson M., Feldman S.A., Maric I., Raffeld M., Nathan D.A., Lanier B.J., et al. Eradication of B-lineage cells and regression of lymphoma in a patient treated with autologous T cells genetically engineered to recognize CD19. Blood. 2010;116:4099–4102. doi: 10.1182/blood-2010-04-281931. - DOI - PMC - PubMed
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
