

Stress tensor and constant pressure simulation for polarizable Gaussian multipole model
- PMID: 35317572
- PMCID: PMC9088672
- DOI: 10.1063/5.0082548
Stress tensor and constant pressure simulation for polarizable Gaussian multipole model
Abstract
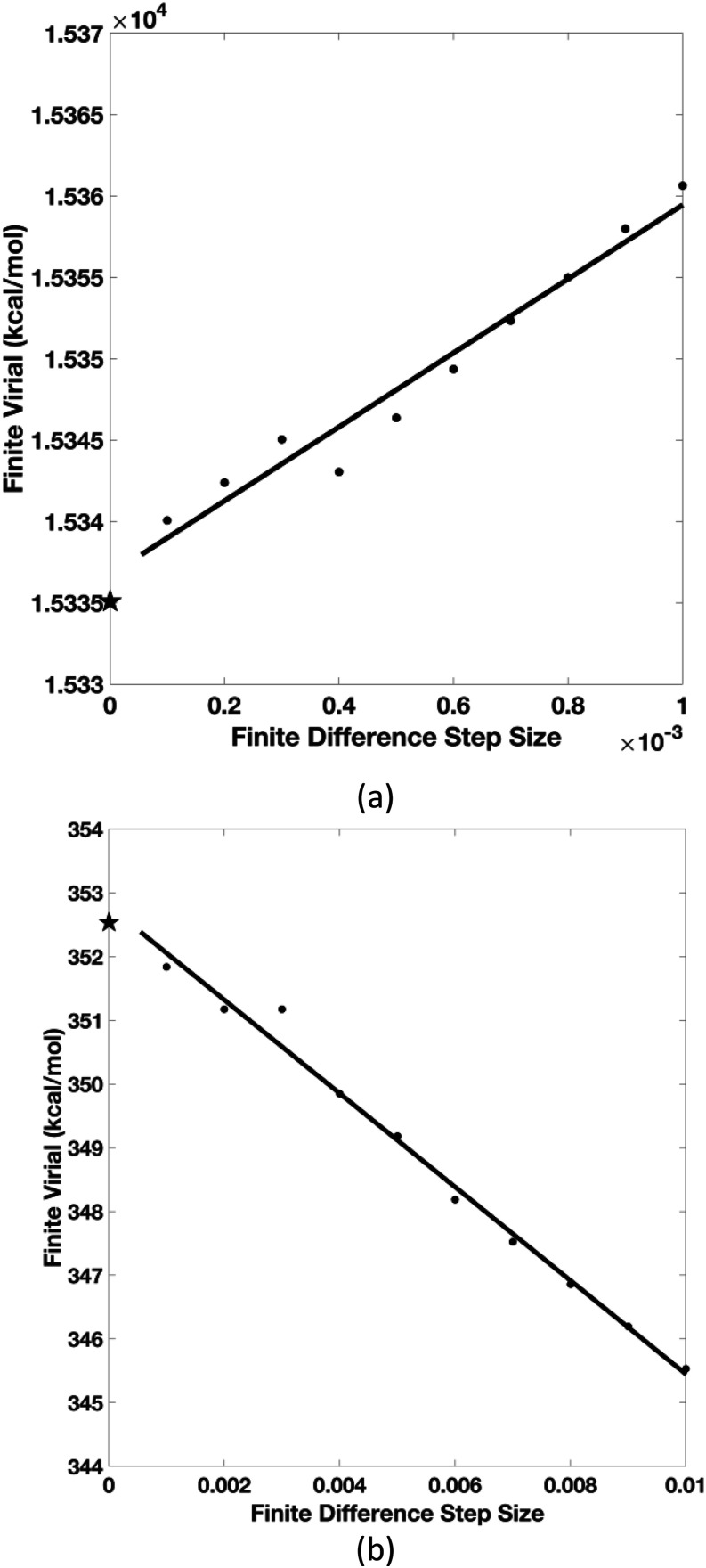
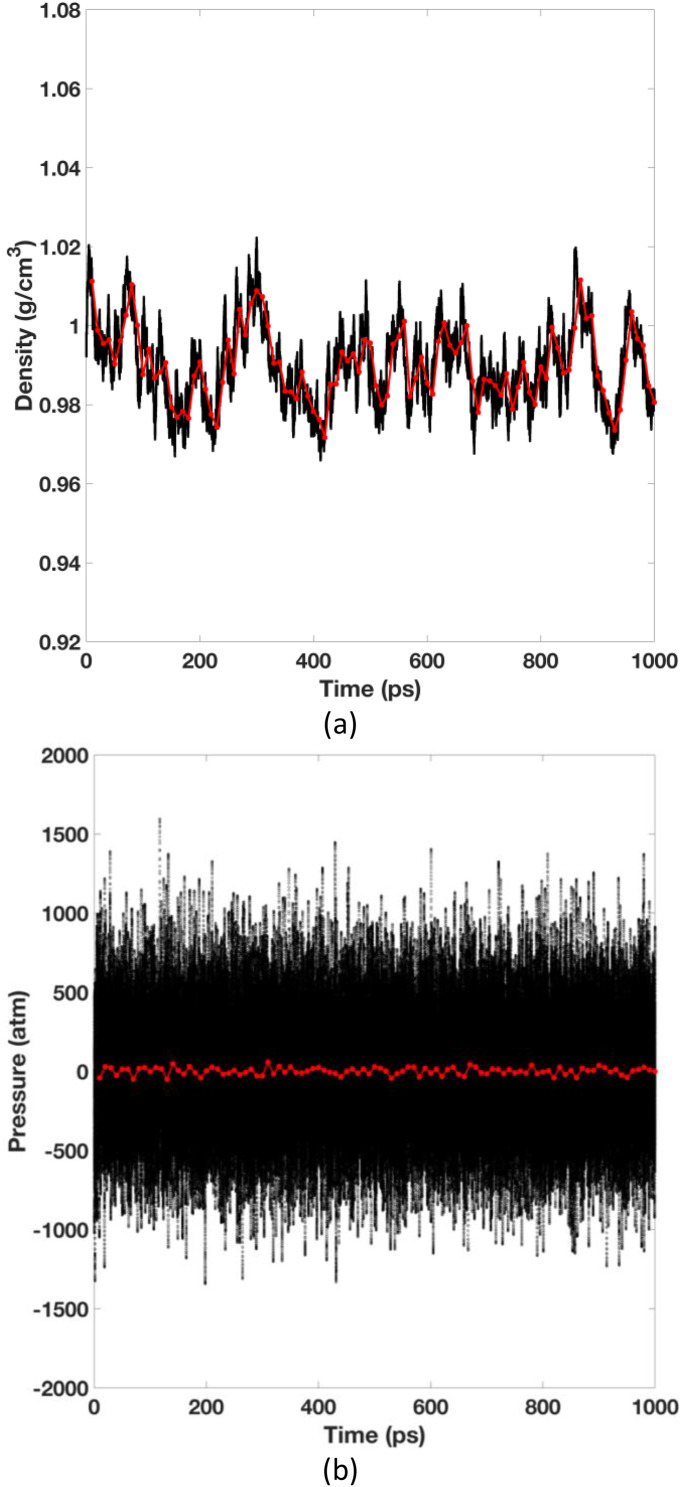
Our previous article has established the theory of molecular dynamics (MD) simulations for systems modeled with the polarizable Gaussian multipole (pGM) electrostatics [Wei et al., J. Chem. Phys. 153(11), 114116 (2020)]. Specifically, we proposed the covalent basis vector framework to define the permanent multipoles and derived closed-form energy and force expressions to facilitate an efficient implementation of pGM electrostatics. In this study, we move forward to derive the pGM internal stress tensor for constant pressure MD simulations with the pGM electrostatics. Three different formulations are presented for the flexible, rigid, and short-range screened systems, respectively. The analytical formulations were implemented in the SANDER program in the Amber package and were first validated with the finite-difference method for two different boxes of pGM water molecules. This is followed by a constant temperature and constant pressure MD simulation for a box of 512 pGM water molecules. Our results show that the simulation system stabilized at a physically reasonable state and maintained the balance with the externally applied pressure. In addition, several fundamental differences were observed between the pGM and classic point charge models in terms of the simulation behaviors, indicating more extensive parameterization is necessary to utilize the pGM electrostatics.
Figures
Similar articles
-
Performance Tuning of Polarizable Gaussian Multipole Model in Molecular Dynamics Simulations.J Chem Theory Comput. 2025 Jan 28;21(2):847-858. doi: 10.1021/acs.jctc.4c01368. Epub 2025 Jan 8. J Chem Theory Comput. 2025. PMID: 39772516 Free PMC article.
-
Efficient formulation of polarizable Gaussian multipole electrostatics for biomolecular simulations.J Chem Phys. 2020 Sep 21;153(11):114116. doi: 10.1063/5.0019560. J Chem Phys. 2020. PMID: 32962395 Free PMC article.
-
Isotropic Periodic Sum for Polarizable Gaussian Multipole Model.J Chem Theory Comput. 2025 Apr 22;21(8):4040-4050. doi: 10.1021/acs.jctc.5c00123. Epub 2025 Apr 7. J Chem Theory Comput. 2025. PMID: 40194962
-
Transferability of the Electrostatic Parameters of the Polarizable Gaussian Multipole Model.J Chem Theory Comput. 2023 Feb 14;19(3):924-941. doi: 10.1021/acs.jctc.2c01048. Epub 2023 Jan 25. J Chem Theory Comput. 2023. PMID: 36696564 Free PMC article.
-
Biomolecular force fields: where have we been, where are we now, where do we need to go and how do we get there?J Comput Aided Mol Des. 2019 Feb;33(2):133-203. doi: 10.1007/s10822-018-0111-4. Epub 2018 Nov 30. J Comput Aided Mol Des. 2019. PMID: 30506158 Review.
Cited by
-
Performance Tuning of Polarizable Gaussian Multipole Model in Molecular Dynamics Simulations.J Chem Theory Comput. 2025 Jan 28;21(2):847-858. doi: 10.1021/acs.jctc.4c01368. Epub 2025 Jan 8. J Chem Theory Comput. 2025. PMID: 39772516 Free PMC article.
-
Development of First-in-Class Dual Sirt2/HDAC6 Inhibitors as Molecular Tools for Dual Inhibition of Tubulin Deacetylation.J Med Chem. 2023 Nov 9;66(21):14787-14814. doi: 10.1021/acs.jmedchem.3c01385. Epub 2023 Oct 30. J Med Chem. 2023. PMID: 37902787 Free PMC article.
-
PyRESP: A Program for Electrostatic Parameterizations of Additive and Induced Dipole Polarizable Force Fields.J Chem Theory Comput. 2022 Jun 14;18(6):3654-3670. doi: 10.1021/acs.jctc.2c00230. Epub 2022 May 10. J Chem Theory Comput. 2022. PMID: 35537209 Free PMC article.
-
TinkerModeller: An Efficient Tool for Building Biological Systems in Tinker Simulations.J Chem Theory Comput. 2025 Mar 11;21(5):2712-2722. doi: 10.1021/acs.jctc.4c01463. Epub 2025 Feb 25. J Chem Theory Comput. 2025. PMID: 39999350 Free PMC article.
-
Refinement of Atomic Polarizabilities for a Polarizable Gaussian Multipole Force Field with Simultaneous Considerations of Both Molecular Polarizability Tensors and In-Solution Electrostatic Potentials.J Chem Inf Model. 2025 Feb 10;65(3):1428-1440. doi: 10.1021/acs.jcim.4c02175. Epub 2025 Jan 26. J Chem Inf Model. 2025. PMID: 39865620 Free PMC article.
References
-
- Burkert U. and Allinger N., Molecular Mechanics (American Chemical Society, Washington, DC, 1982).
-
- Jorgensen W. L., Chandrasekhar J., Madura J. D., Impey R. W., and Klein M. L., “Comparison of simple potential functions for simulating liquid water,” J. Chem. Phys. 79(2), 926–935 (1983).10.1063/1.445869 - DOI
-
- Berendsen H. J. C., Postma J. P. M., van Gunsteren W. F., and Hermans J., “Interaction models for water in relation to protein hydration,” in Intermolecular Forces (Springer, 1981), pp. 331–342.
-
- Mahoney M. W. and Jorgensen W. L., “A five-site model for liquid water and the reproduction of the density anomaly by rigid, non-polarizable potential functions,” J. Chem. Phys. 112(20), 8910–8922 (2000).10.1063/1.481505 - DOI