

A comparative methylome analysis reveals conservation and divergence of DNA methylation patterns and functions in vertebrates
- PMID: 35317801
- PMCID: PMC8941758
- DOI: 10.1186/s12915-022-01270-x
A comparative methylome analysis reveals conservation and divergence of DNA methylation patterns and functions in vertebrates
Abstract
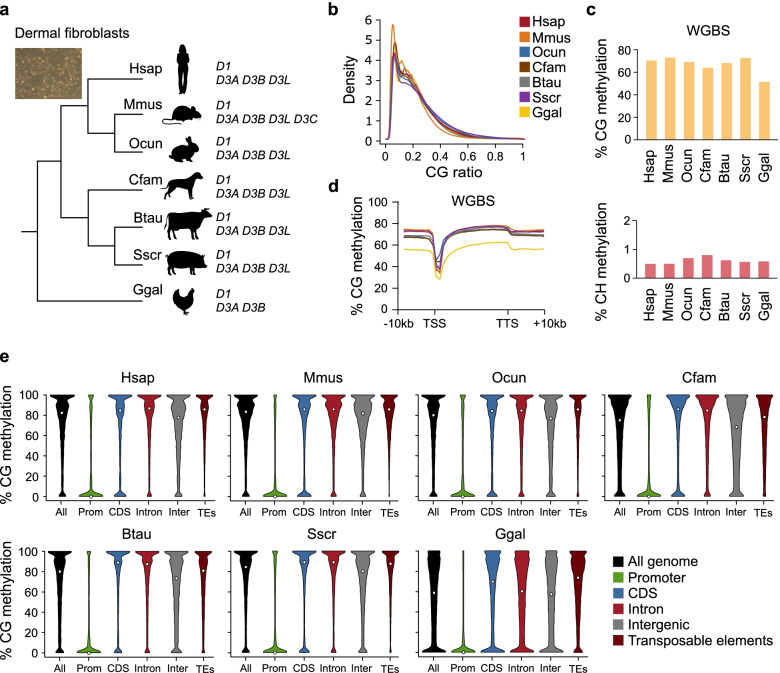
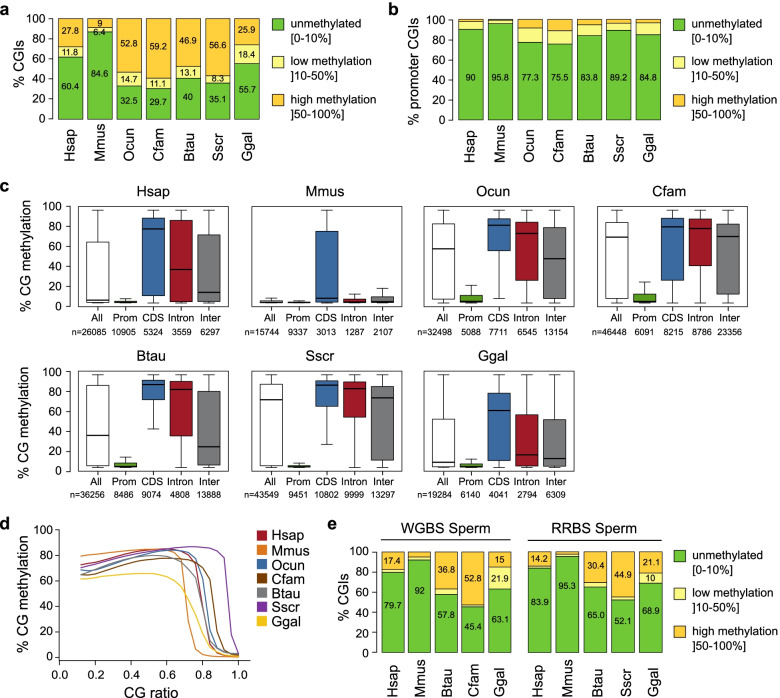
Background: Cytosine DNA methylation is a heritable epigenetic mark present in most eukaryotic groups. While the patterns and functions of DNA methylation have been extensively studied in mouse and human, their conservation in other vertebrates remains poorly explored. In this study, we interrogated the distribution and function of DNA methylation in primary fibroblasts of seven vertebrate species including bio-medical models and livestock species (human, mouse, rabbit, dog, cow, pig, and chicken).
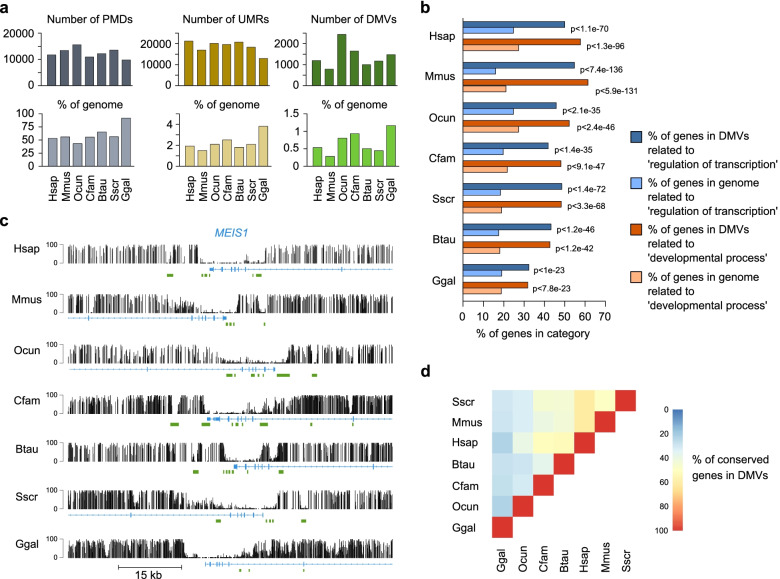
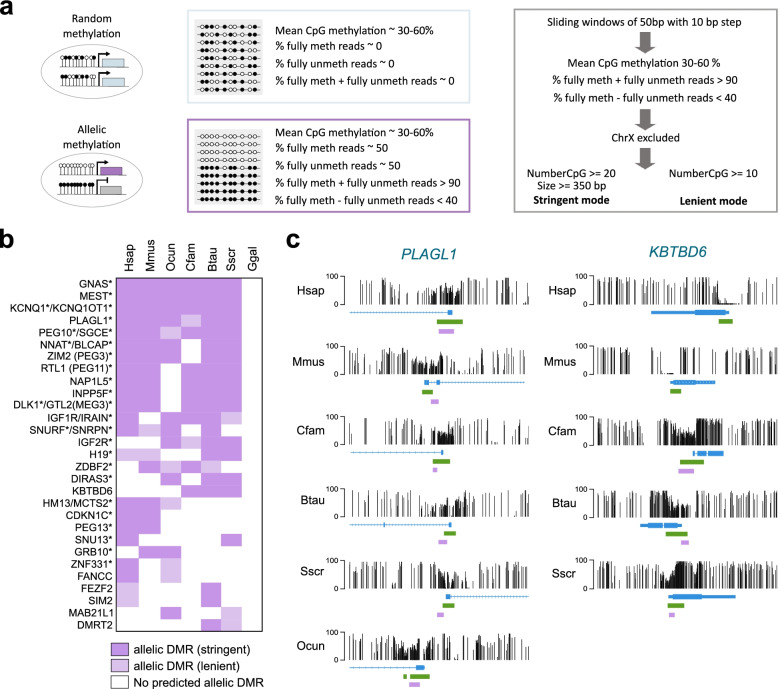
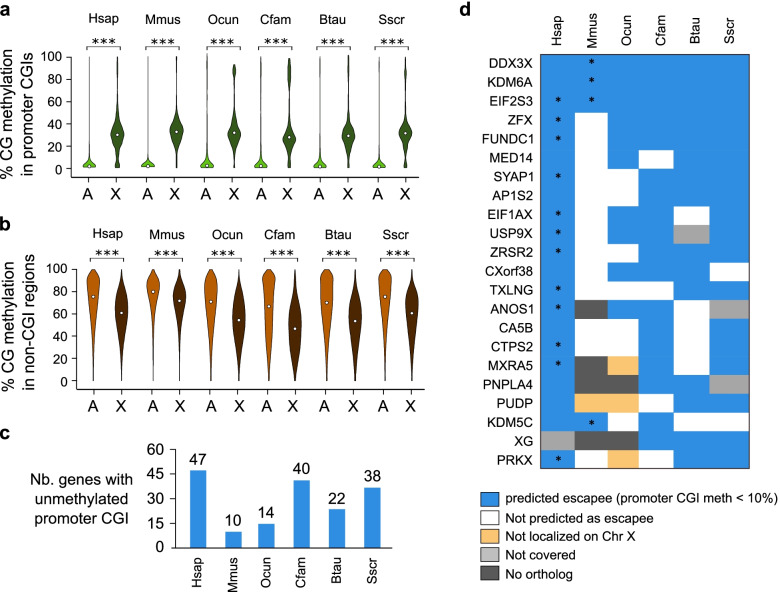
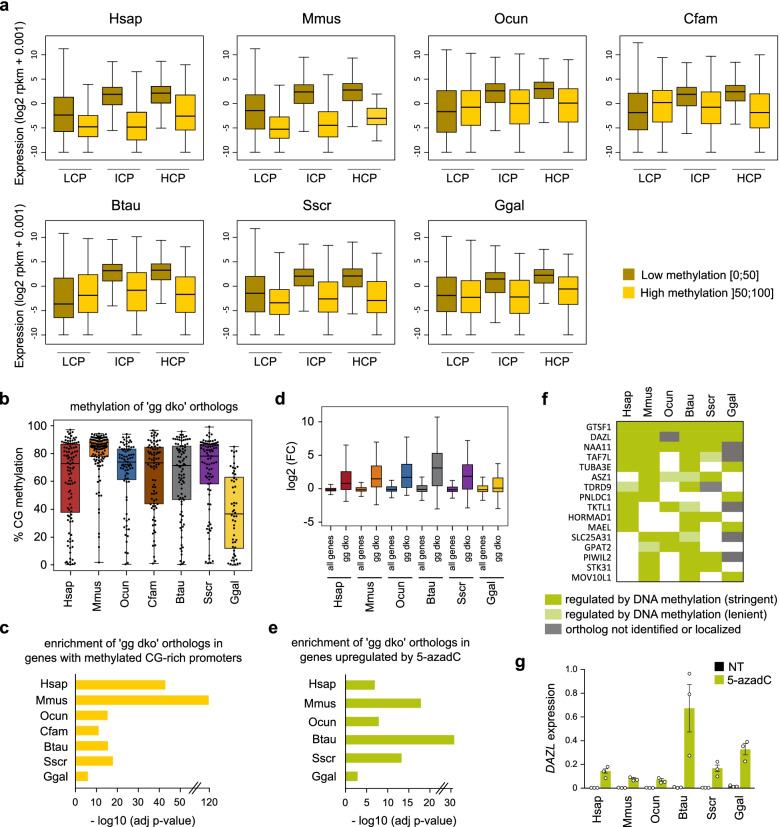
Results: Our data highlight both divergence and conservation of DNA methylation patterns and functions. We show that the chicken genome is hypomethylated compared to other vertebrates. Furthermore, compared to mouse, other species show a higher frequency of methylation of CpG-rich DNA. We reveal the conservation of large unmethylated valleys and patterns of DNA methylation associated with X-chromosome inactivation through vertebrate evolution and make predictions of conserved sets of imprinted genes across mammals. Finally, using chemical inhibition of DNA methylation, we show that the silencing of germline genes and endogenous retroviruses (ERVs) are conserved functions of DNA methylation in vertebrates.
Conclusions: Our data highlight conserved properties of DNA methylation in vertebrate genomes but at the same time point to differences between mouse and other vertebrate species.
Keywords: 5mC; CpG island; DNA methylation; Genomic imprinting; Germline genes; Vertebrates.
© 2022. The Author(s).
Conflict of interest statement
The authors declare that they have no competing interests.
Figures
References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
