

S6K1-mediated phosphorylation of PDK1 impairs AKT kinase activity and oncogenic functions
- PMID: 35318320
- PMCID: PMC8941131
- DOI: 10.1038/s41467-022-28910-8
S6K1-mediated phosphorylation of PDK1 impairs AKT kinase activity and oncogenic functions
Abstract
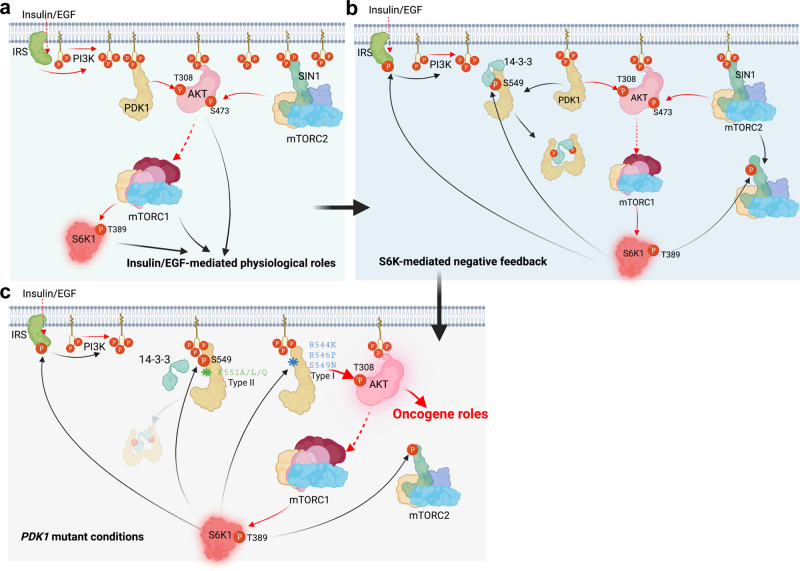
Functioning as a master kinase, 3-phosphoinositide-dependent protein kinase 1 (PDK1) plays a fundamental role in phosphorylating and activating protein kinases A, B and C (AGC) family kinases, including AKT. However, upstream regulation of PDK1 remains largely elusive. Here we report that ribosomal protein S6 kinase beta 1 (S6K1), a member of AGC kinases and downstream target of mechanistic target of rapamycin complex 1 (mTORC1), directly phosphorylates PDK1 at its pleckstrin homology (PH) domain, and impairs PDK1 interaction with and activation of AKT. Mechanistically, S6K1-mediated phosphorylation of PDK1 augments its interaction with 14-3-3 adaptor protein and homo-dimerization, subsequently dissociating PDK1 from phosphatidylinositol 3,4,5 triphosphate (PIP3) and retarding its interaction with AKT. Pathologically, tumor patient-associated PDK1 mutations, either attenuating S6K1-mediated PDK1 phosphorylation or impairing PDK1 interaction with 14-3-3, result in elevated AKT kinase activity and oncogenic functions. Taken together, our findings not only unravel a delicate feedback regulation of AKT signaling via S6K1-mediated PDK1 phosphorylation, but also highlight the potential strategy to combat mutant PDK1-driven cancers.
© 2022. The Author(s).
Conflict of interest statement
W.W. is a co-founder and consultant for the ReKindle Therapeutics. Other authors declare no competing interests.
Figures






References
-
- Blume-Jensen P, Hunter T. Oncogenic kinase signalling. Nature. 2001;411:355–365. - PubMed
-
- Pearce LR, Komander D, Alessi DR. The nuts and bolts of AGC protein kinases. Nat. Rev. Mol. Cell Biol. 2010;11:9–22. - PubMed
-
- Mora A, Komander D, van Aalten DM, Alessi DR. PDK1, the master regulator of AGC kinase signal transduction. Semin Cell Dev. Biol. 2004;15:161–170. - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Miscellaneous
