

Combinatorial optimization of mRNA structure, stability, and translation for RNA-based therapeutics
- PMID: 35318324
- PMCID: PMC8940940
- DOI: 10.1038/s41467-022-28776-w
Combinatorial optimization of mRNA structure, stability, and translation for RNA-based therapeutics
Abstract
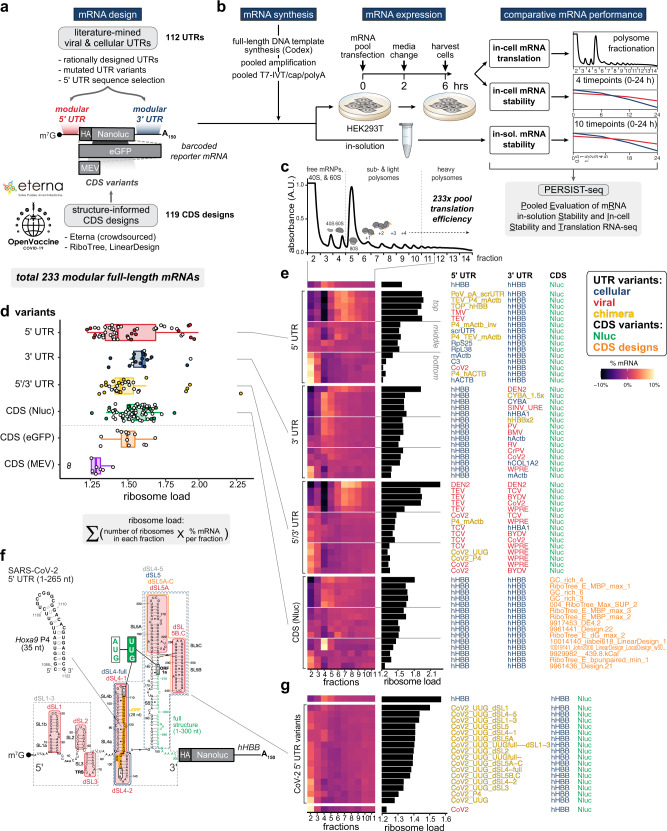
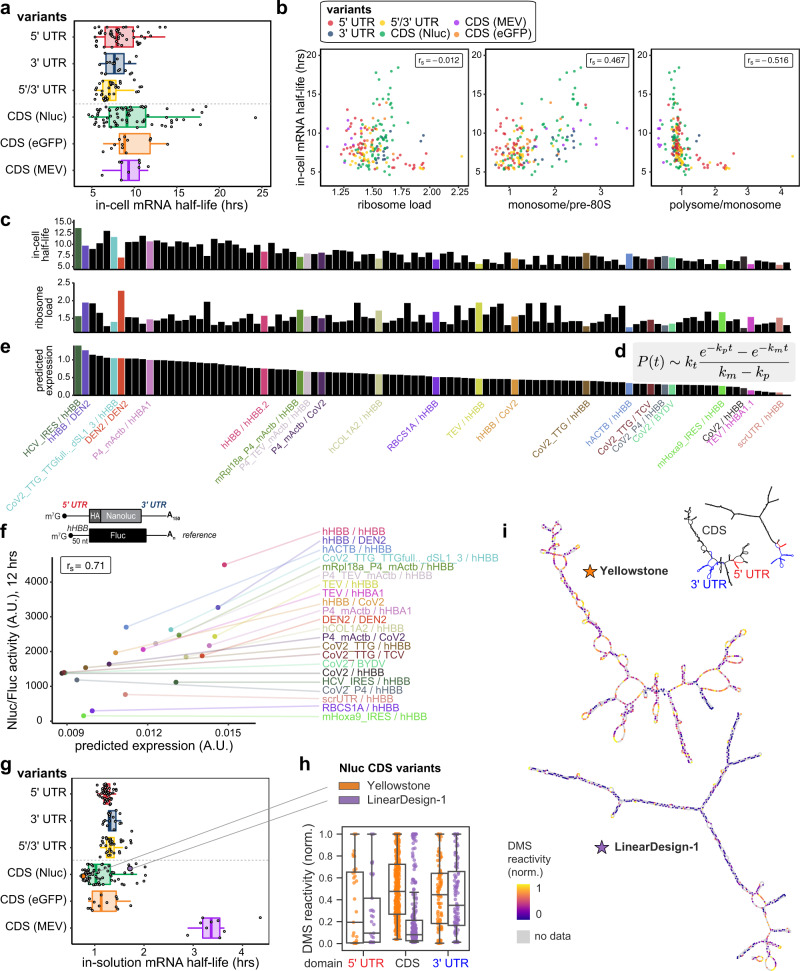
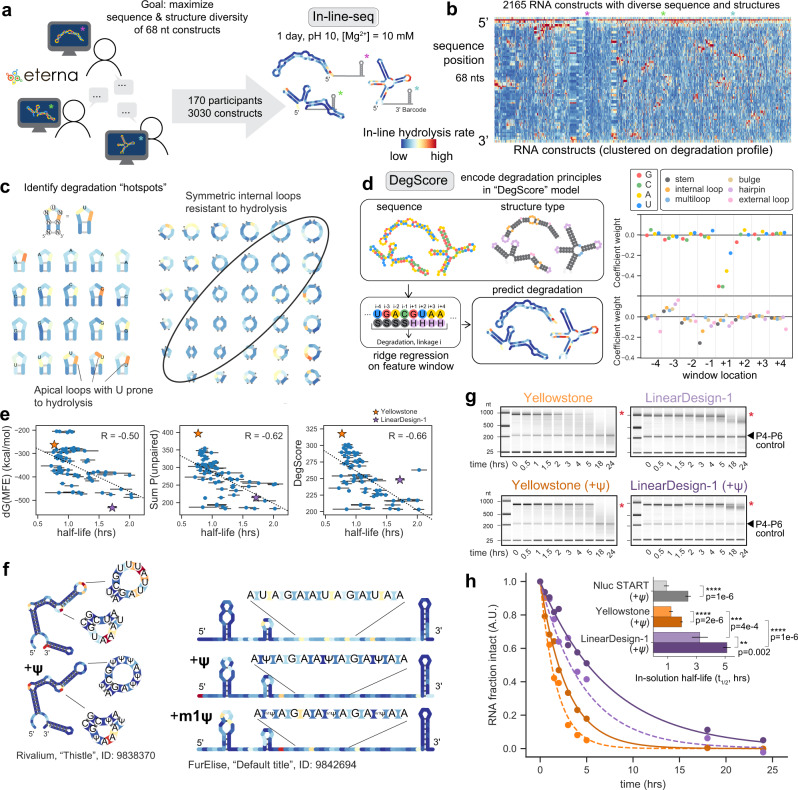
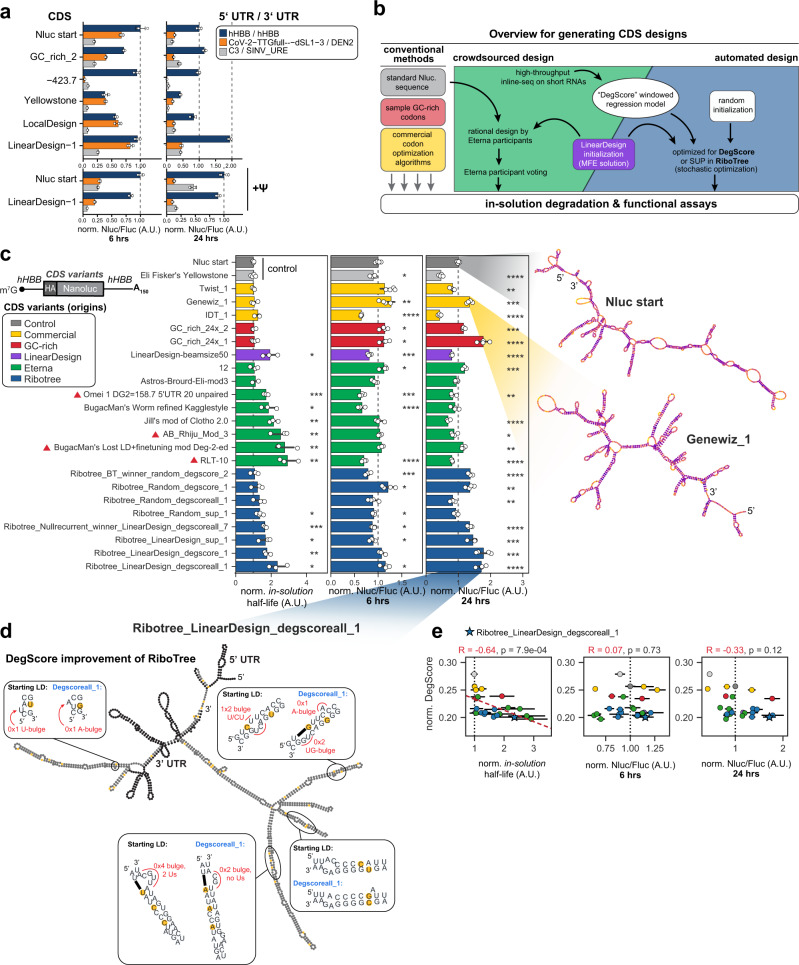
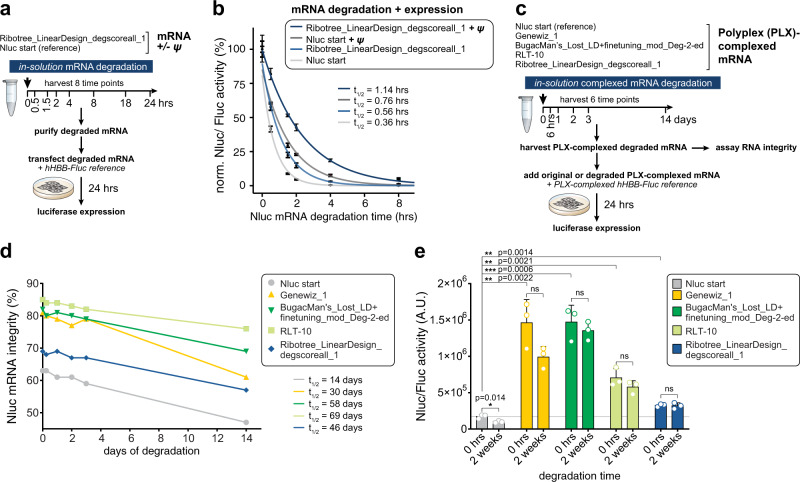
Therapeutic mRNAs and vaccines are being developed for a broad range of human diseases, including COVID-19. However, their optimization is hindered by mRNA instability and inefficient protein expression. Here, we describe design principles that overcome these barriers. We develop an RNA sequencing-based platform called PERSIST-seq to systematically delineate in-cell mRNA stability, ribosome load, as well as in-solution stability of a library of diverse mRNAs. We find that, surprisingly, in-cell stability is a greater driver of protein output than high ribosome load. We further introduce a method called In-line-seq, applied to thousands of diverse RNAs, that reveals sequence and structure-based rules for mitigating hydrolytic degradation. Our findings show that highly structured "superfolder" mRNAs can be designed to improve both stability and expression with further enhancement through pseudouridine nucleoside modification. Together, our study demonstrates simultaneous improvement of mRNA stability and protein expression and provides a computational-experimental platform for the enhancement of mRNA medicines.
© 2022. The Author(s).
Conflict of interest statement
Stanford University has submitted provisional patent applications related to use of the
Figures
Update of
-
Combinatorial optimization of mRNA structure, stability, and translation for RNA-based therapeutics.bioRxiv [Preprint]. 2021 Mar 30:2021.03.29.437587. doi: 10.1101/2021.03.29.437587. bioRxiv. 2021. Update in: Nat Commun. 2022 Mar 22;13(1):1536. doi: 10.1038/s41467-022-28776-w. PMID: 33821271 Free PMC article. Updated. Preprint.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
