

mTORC2 suppresses cell death induced by hypo-osmotic stress by promoting sphingomyelin transport
- PMID: 35319770
- PMCID: PMC8952684
- DOI: 10.1083/jcb.202106160
mTORC2 suppresses cell death induced by hypo-osmotic stress by promoting sphingomyelin transport
Abstract
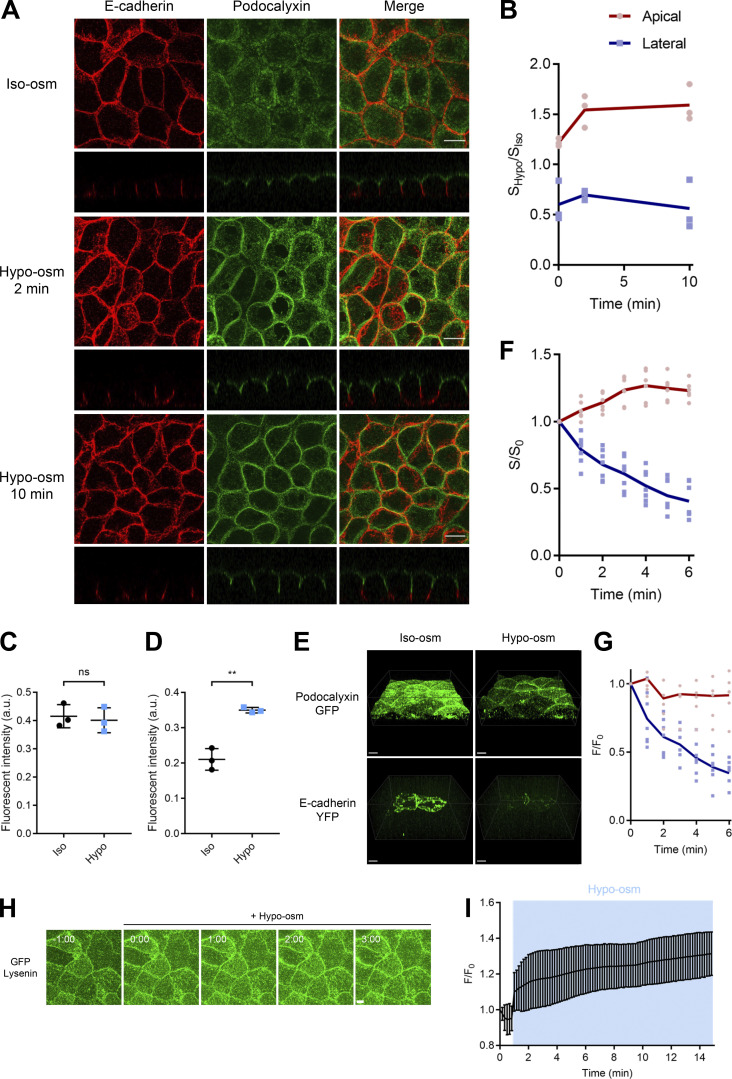
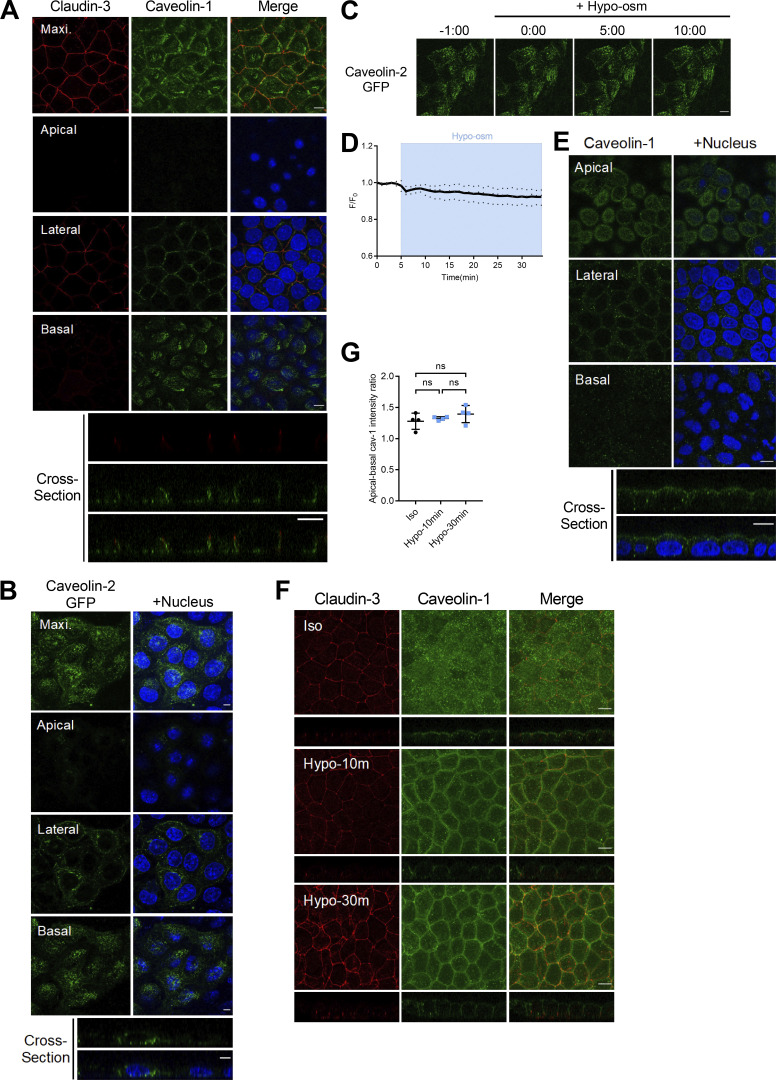
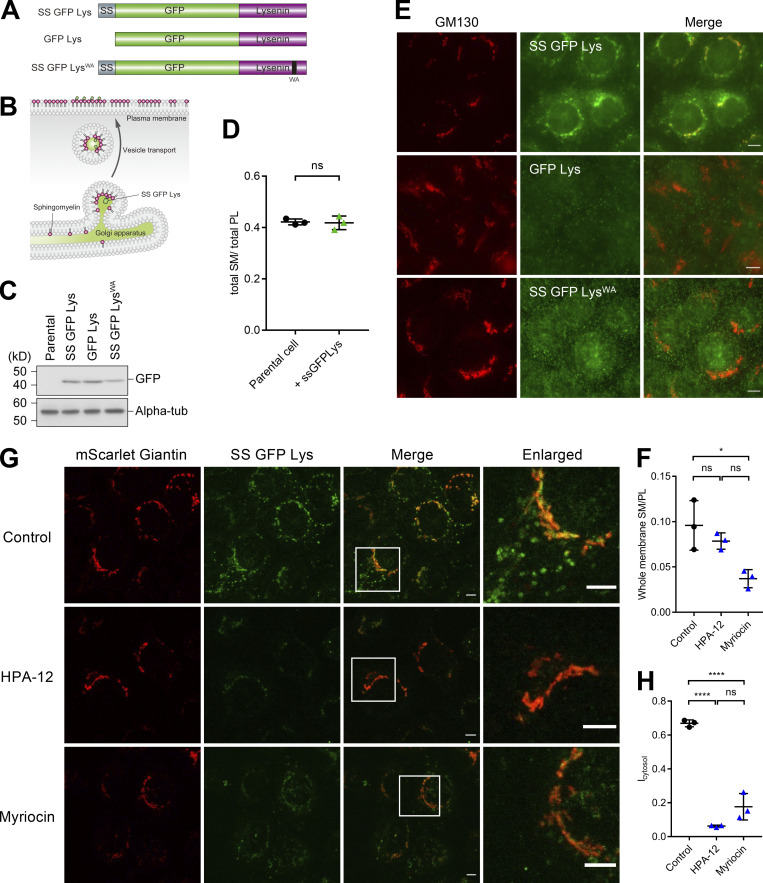
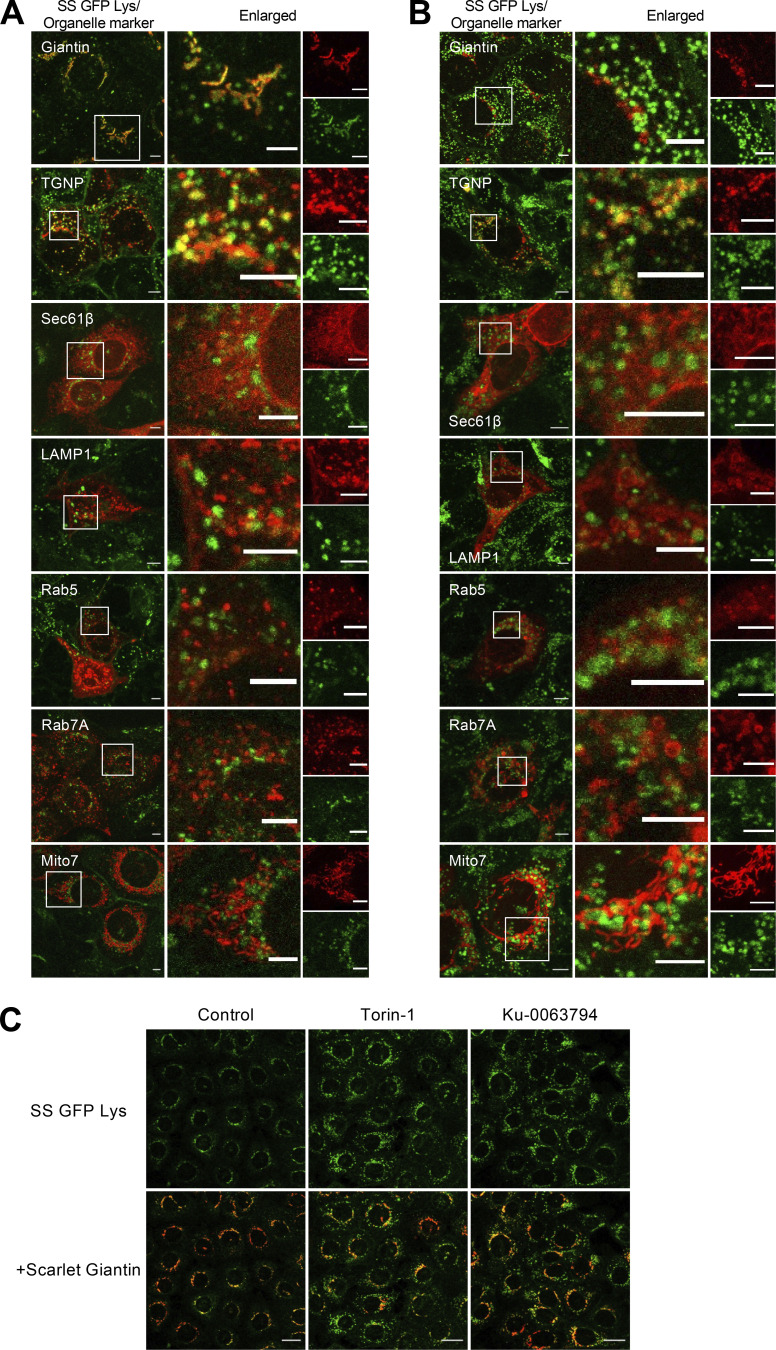
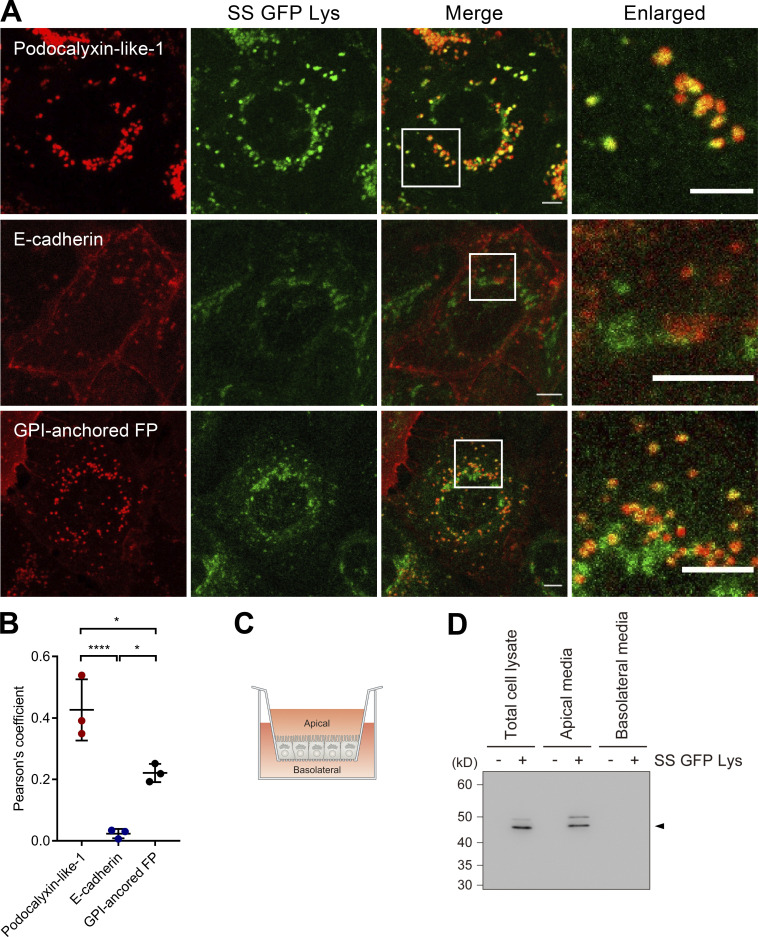
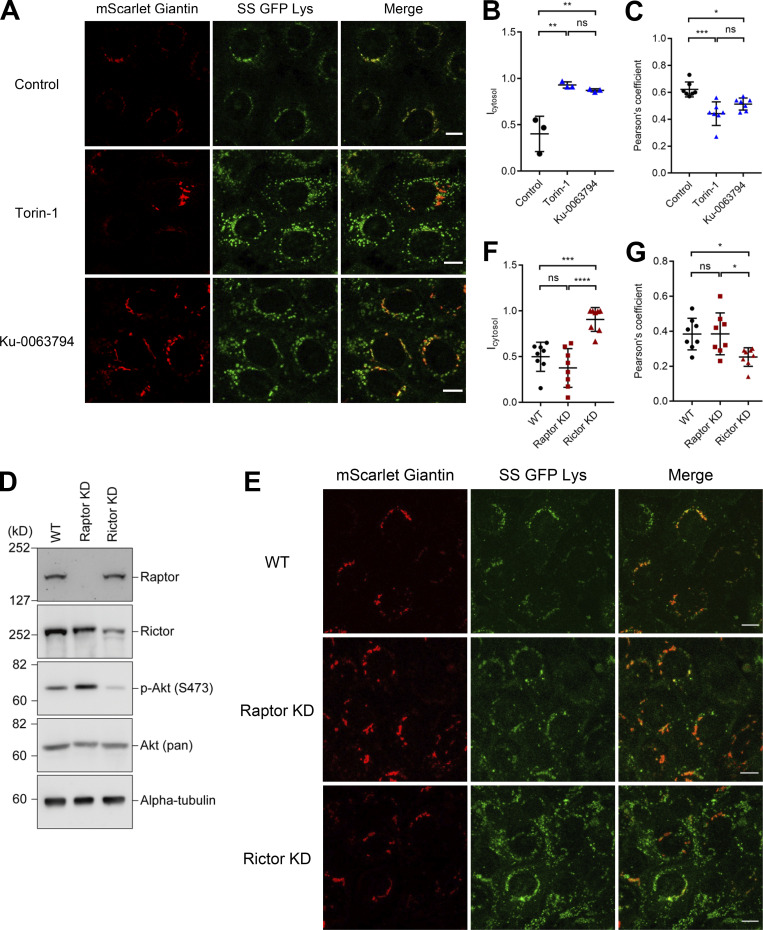
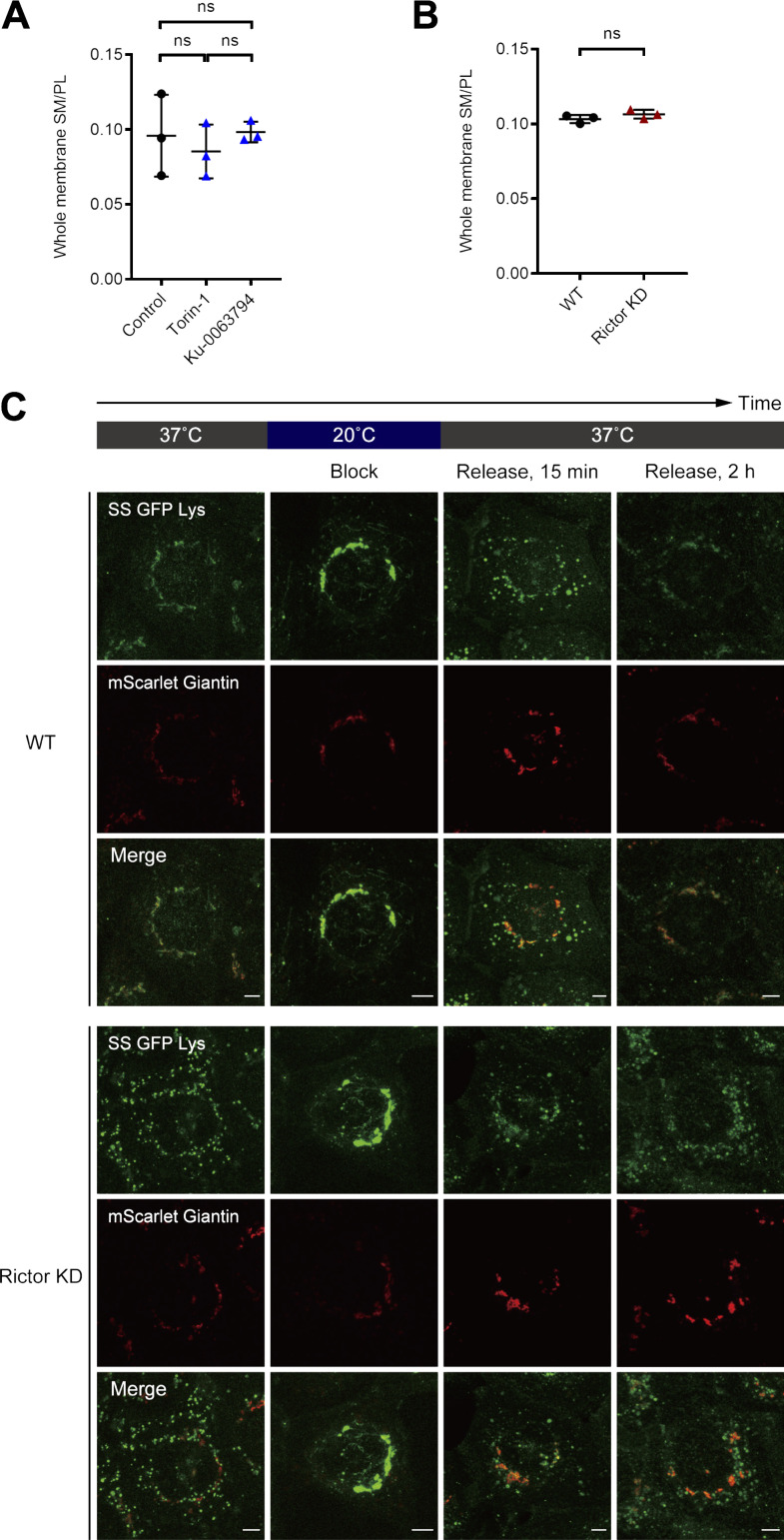
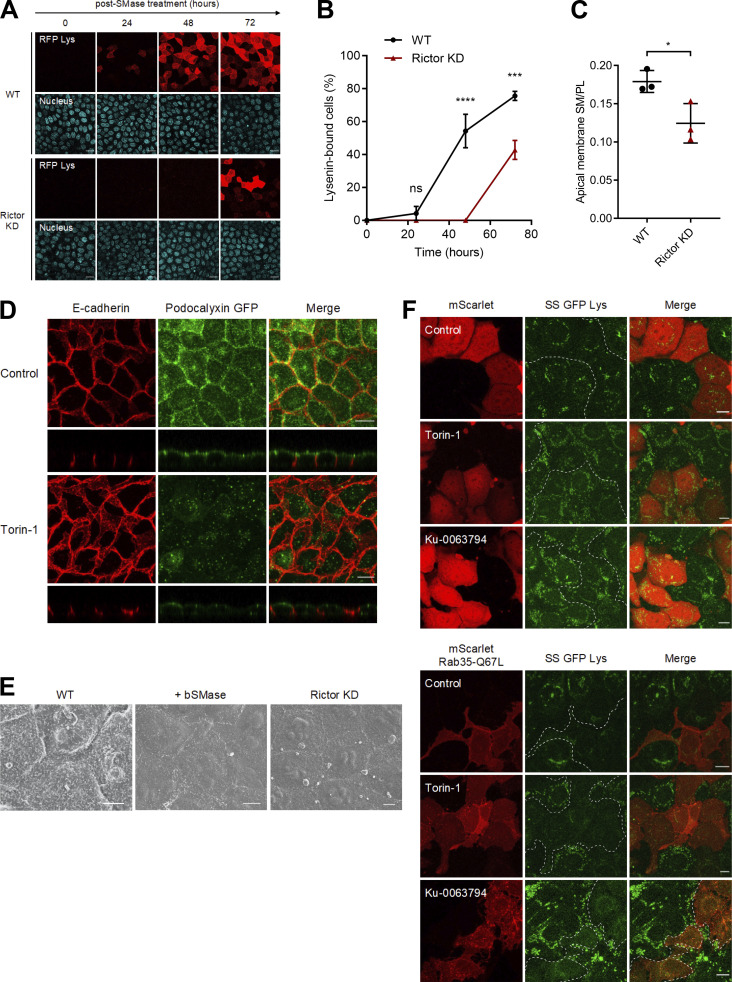
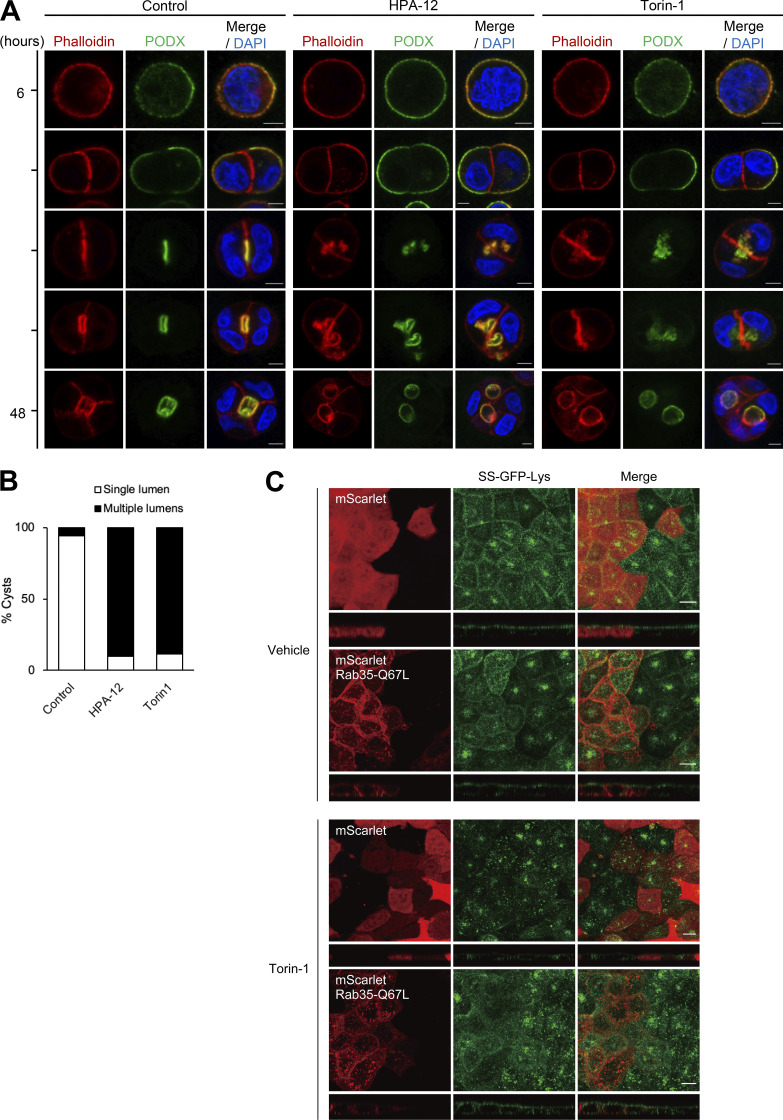
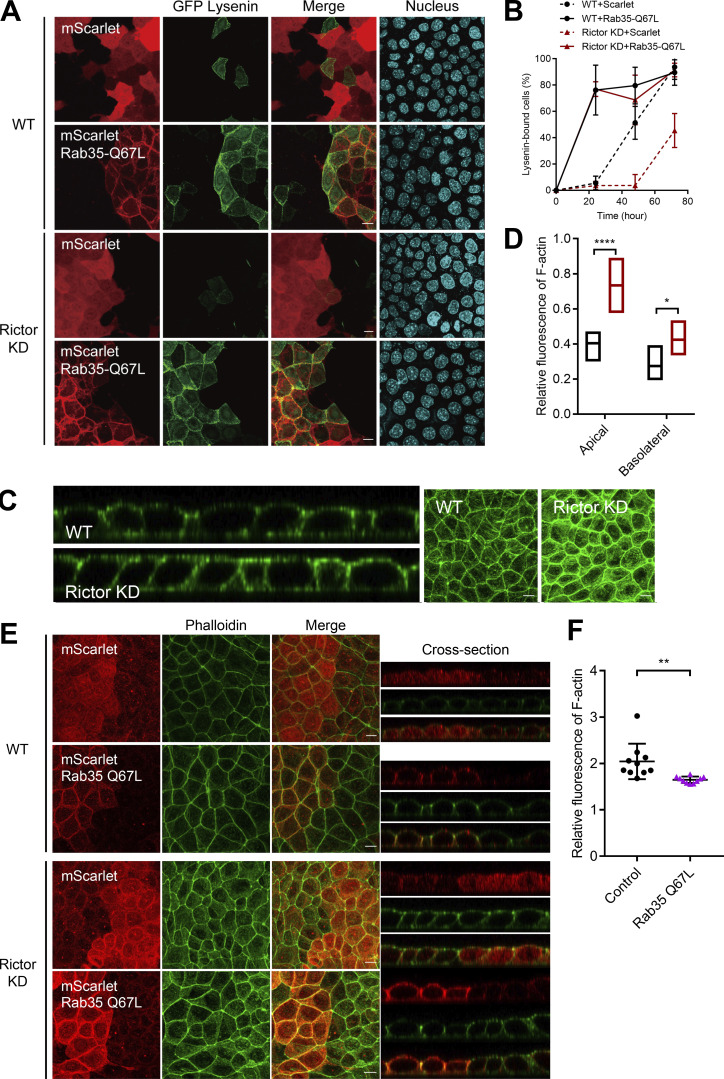
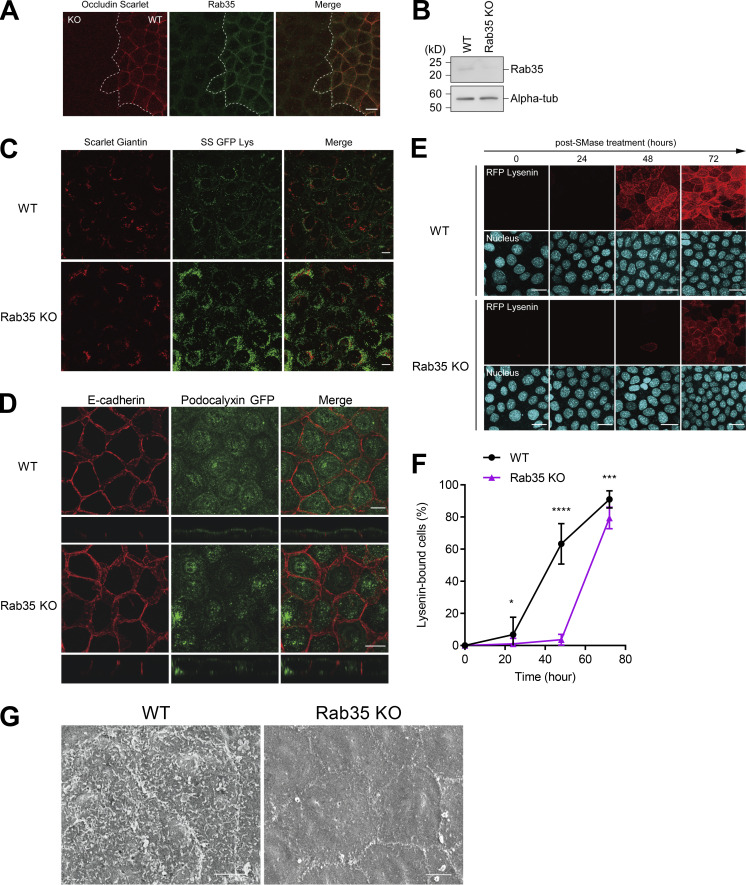
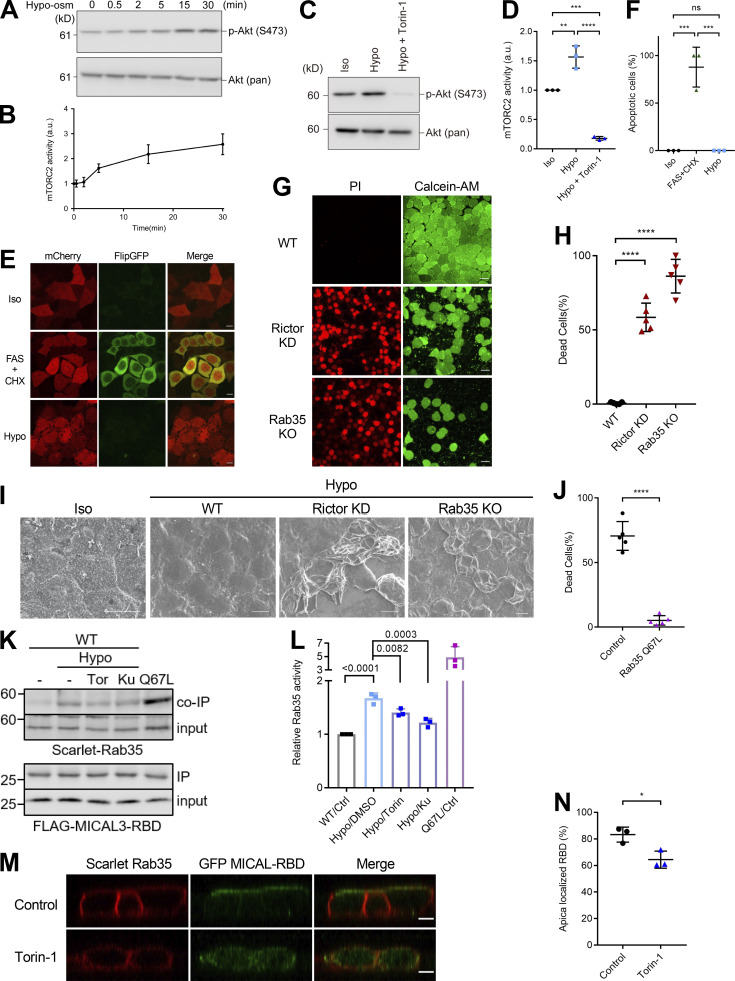
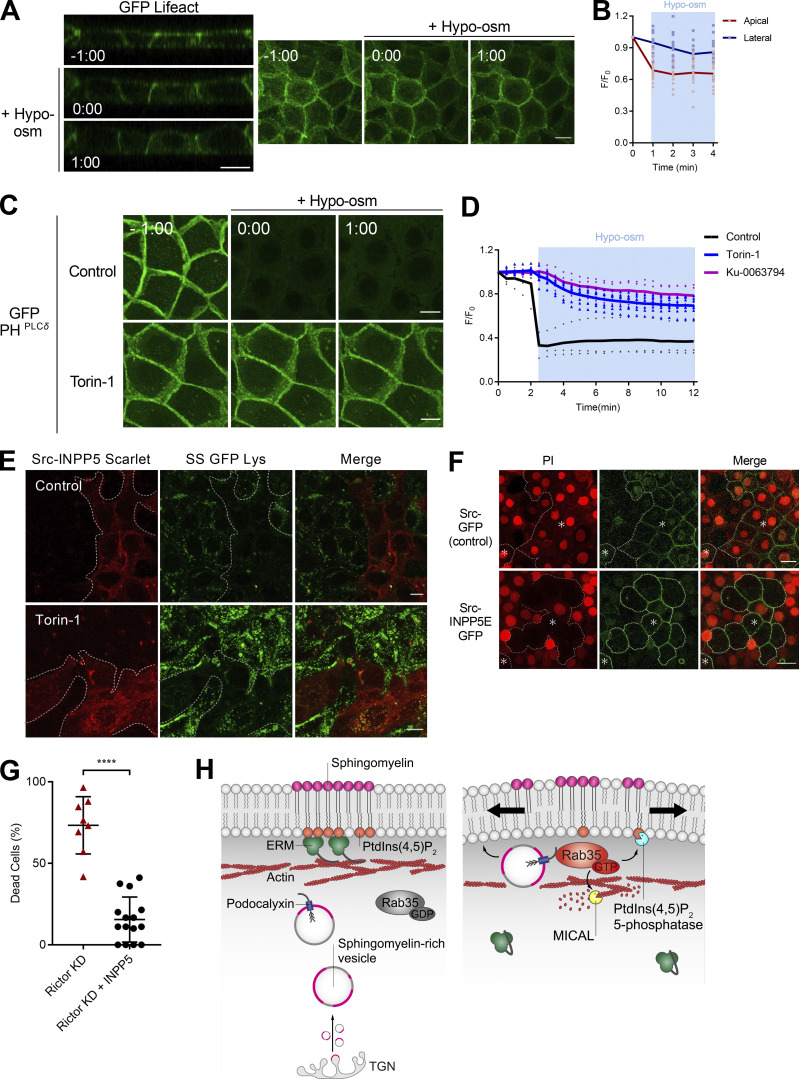
Epithelial cells are constantly exposed to osmotic stress. The influx of water molecules into the cell in a hypo-osmotic environment increases plasma membrane tension as it rapidly expands. Therefore, the plasma membrane must be supplied with membrane lipids since expansion beyond its elastic limit will cause the cell to rupture. However, the molecular mechanism to maintain a constant plasma membrane tension is not known. In this study, we found that the apical membrane selectively expands when epithelial cells are exposed to hypo-osmotic stress. This requires the activation of mTORC2, which enhances the transport of secretory vesicles containing sphingomyelin, the major lipid of the apical membrane. We further show that the mTORC2-Rab35 axis plays an essential role in the defense against hypotonic stress by promoting the degradation of the actin cortex through the up-regulation of PI(4,5)P2 metabolism, which facilitates the apical tethering of sphingomyelin-loaded vesicles to relieve plasma membrane tension.
© 2022 Ono et al.
Figures
Similar articles
-
Plasma membrane protein OsMCA1 is involved in regulation of hypo-osmotic shock-induced Ca2+ influx and modulates generation of reactive oxygen species in cultured rice cells.BMC Plant Biol. 2012 Jan 23;12:11. doi: 10.1186/1471-2229-12-11. BMC Plant Biol. 2012. PMID: 22264357 Free PMC article.
-
Decrease in plasma membrane tension triggers PtdIns(4,5)P2 phase separation to inactivate TORC2.Nat Cell Biol. 2018 Sep;20(9):1043-1051. doi: 10.1038/s41556-018-0150-z. Epub 2018 Aug 27. Nat Cell Biol. 2018. PMID: 30154550 Free PMC article.
-
Membrane tension homeostasis of epithelial cells through surface area regulation in response to osmotic stress.Biochim Biophys Acta. 2013 Mar;1833(3):712-22. doi: 10.1016/j.bbamcr.2012.11.006. Epub 2012 Nov 23. Biochim Biophys Acta. 2013. PMID: 23178740
-
Eisosomes at the intersection of TORC1 and TORC2 regulation.Traffic. 2019 Aug;20(8):543-551. doi: 10.1111/tra.12651. Epub 2019 May 30. Traffic. 2019. PMID: 31038844 Free PMC article. Review.
-
Regulation of the transbilayer movement of diacylglycerol in the plasma membrane.Biochimie. 2014 Dec;107 Pt A:43-50. doi: 10.1016/j.biochi.2014.09.014. Epub 2014 Sep 18. Biochimie. 2014. PMID: 25241257 Review.
Cited by
-
CRISPRi screening identifies PIKfyve as a co-therapeutic target for obinutuzumab.Clin Transl Med. 2025 May;15(5):e70333. doi: 10.1002/ctm2.70333. Clin Transl Med. 2025. PMID: 40342289 Free PMC article. No abstract available.
-
The mTORC2 signaling network: targets and cross-talks.Biochem J. 2024 Jan 25;481(2):45-91. doi: 10.1042/BCJ20220325. Biochem J. 2024. PMID: 38270460 Free PMC article. Review.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
