

Assessment of global health risk of antibiotic resistance genes
- PMID: 35322038
- PMCID: PMC8943045
- DOI: 10.1038/s41467-022-29283-8
Assessment of global health risk of antibiotic resistance genes
Abstract
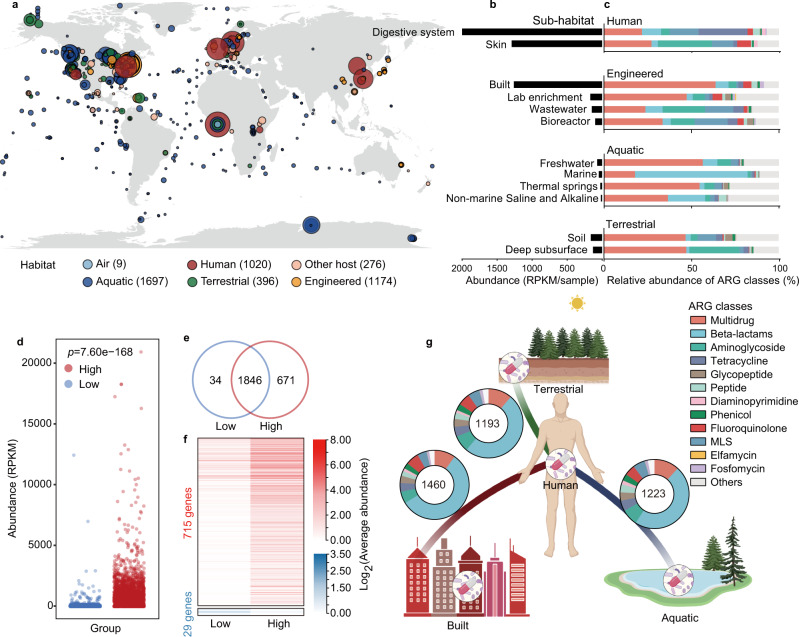
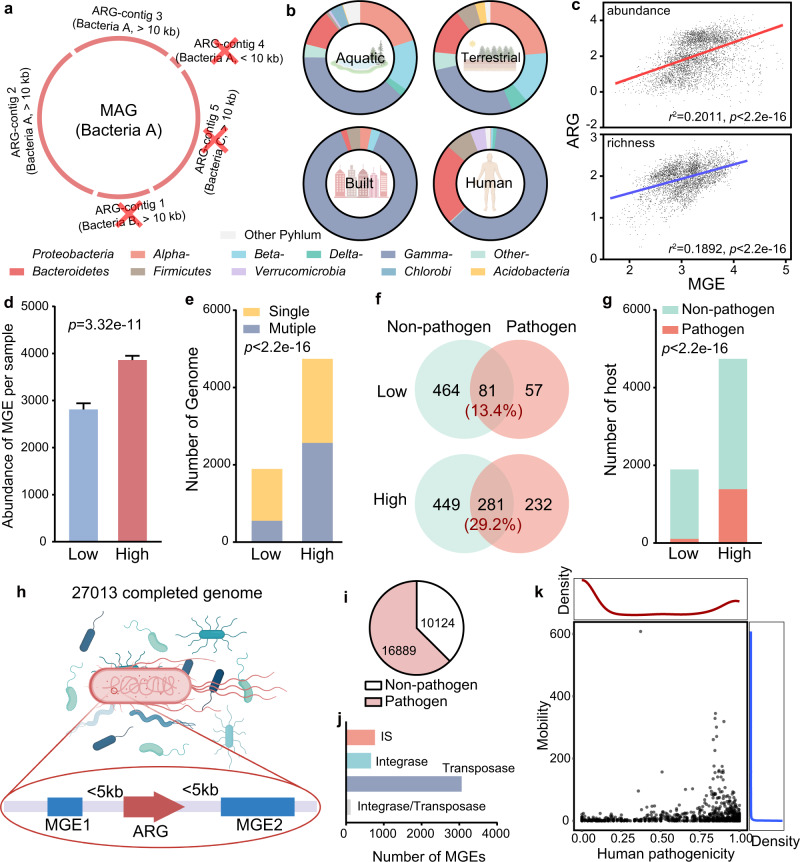
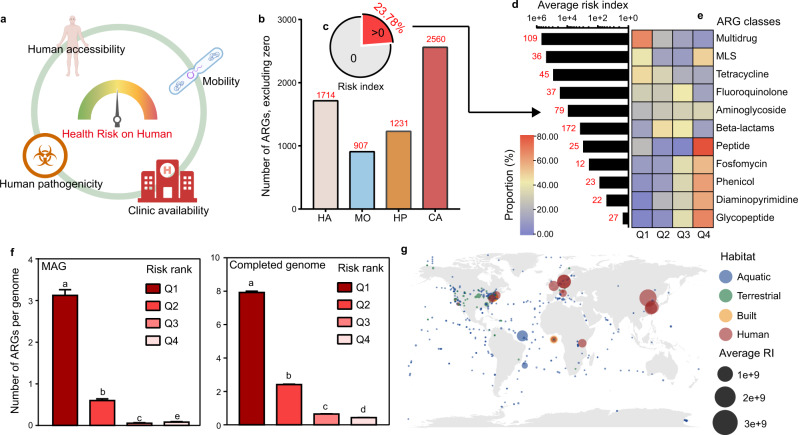
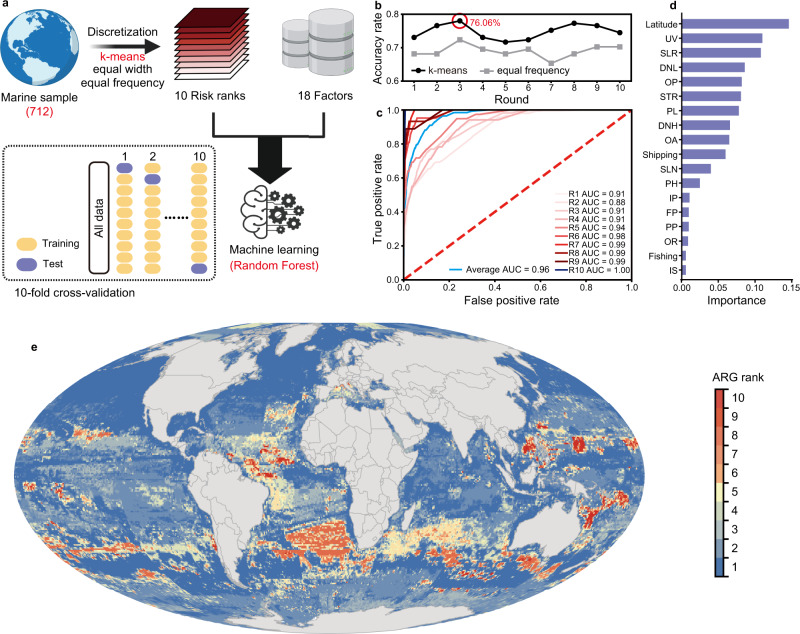
Antibiotic resistance genes (ARGs) have accelerated microbial threats to human health in the last decade. Many genes can confer resistance, but evaluating the relative health risks of ARGs is complex. Factors such as the abundance, propensity for lateral transmission and ability of ARGs to be expressed in pathogens are all important. Here, an analysis at the metagenomic level from various habitats (6 types of habitats, 4572 samples) detects 2561 ARGs that collectively conferred resistance to 24 classes of antibiotics. We quantitatively evaluate the health risk to humans, defined as the risk that ARGs will confound the clinical treatment for pathogens, of these 2561 ARGs by integrating human accessibility, mobility, pathogenicity and clinical availability. Our results demonstrate that 23.78% of the ARGs pose a health risk, especially those which confer multidrug resistance. We also calculate the antibiotic resistance risks of all samples in four main habitats, and with machine learning, successfully map the antibiotic resistance threats in global marine habitats with over 75% accuracy. Our novel method for quantitatively surveilling the health risk of ARGs will help to manage one of the most important threats to human and animal health.
© 2022. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
References
-
- Allen HK, et al. Call of the wild: antibiotic resistance genes in natural environments. Nat. Rev. Microbiol. 2010;8:251–259. - PubMed
-
- Zhu Y-G, et al. Continental-scale pollution of estuaries with antibiotic resistance genes. Nat. Microbiol. 2017;2:16270. - PubMed
-
- Bahram M, et al. Structure and function of the global topsoil microbiome. Nature. 2018;560:233–237. - PubMed
Publication types
MeSH terms
Substances
Associated data
LinkOut - more resources
Full Text Sources
Medical
