

Genomic stratification and differential natural selection signatures among human norovirus genogroup II isolates
- PMID: 35322317
- PMCID: PMC8942050
- DOI: 10.1007/s00705-022-05396-9
Genomic stratification and differential natural selection signatures among human norovirus genogroup II isolates
Abstract
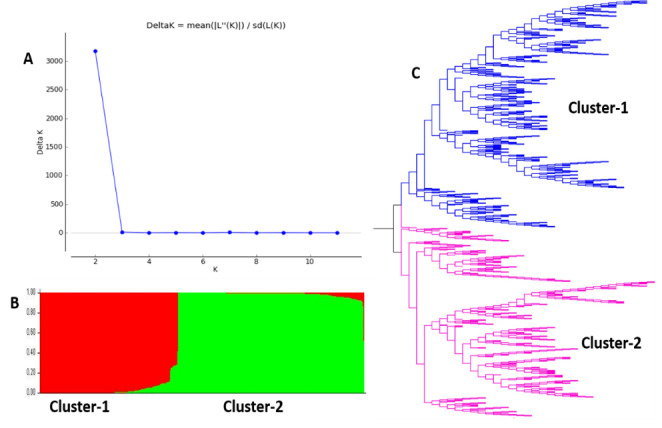
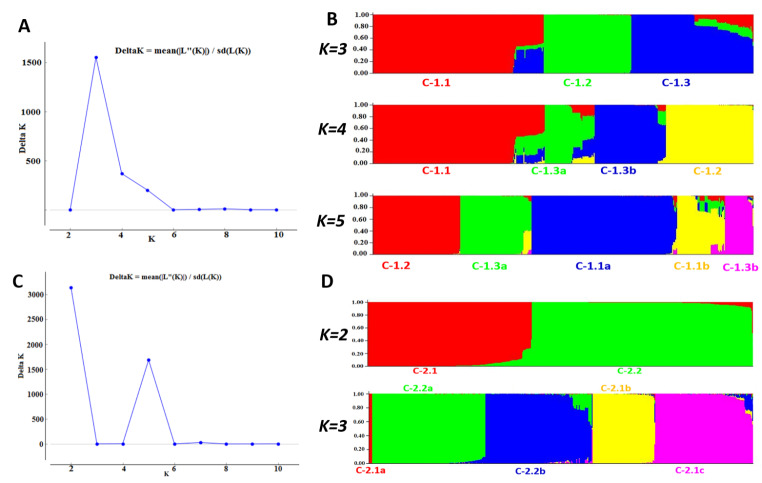
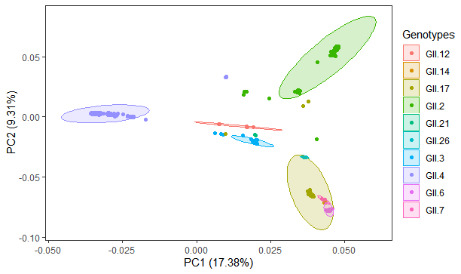
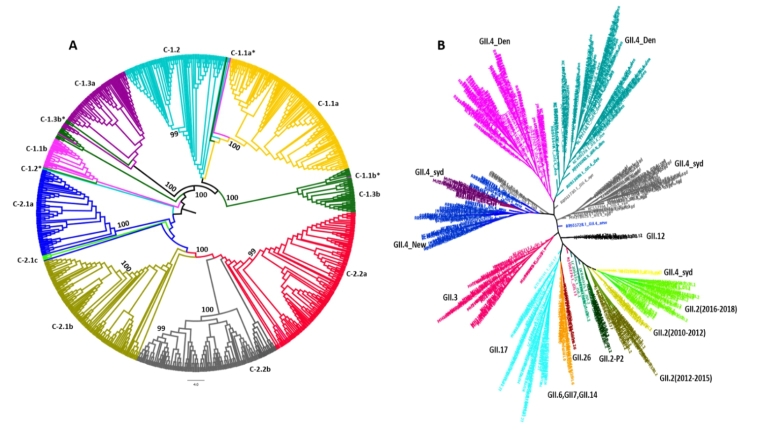
Noroviruses (NoVs), which are members of the family Caliciviridae, are the most common cause of gastroenteritis in humans. Ten NoV genogroups have been reported so far. Of these, genogroup II (GII) is the most prevalent, and it causes serious infections worldwide. The complete genome sequences of NoV GII isolates from different geographical regions were retrieved from the public database. The model-based clustering approach, implemented in the STRUCTURE resource, was employed for assessment of genetic composition. The MEGA X and IQ Tree tools were used for phylogenetic analysis. Genome-wide natural selection analysis was performed using maximum-likelihood-based methods. The demographic features of NoV GII genome sequences were assessed using the BEAST package. All of the NoV GII sequences initially clustered into two main subpopulations at significant K = 2, where the genotype GII.4 samples clearly split from the rest of the genotypes. This indicates a marked genetic distinction between norovirus GII.4 and non-GII.4 samples. Phylogenetic analysis showed the presence of five distinct subclades for genotype GII.2 and seven subclades for GII.4 samples. Several isolates with admixed ancestry were identified that constituted distinct subclusters in the phylogenetic tree. No continental-specific genetic distinctions were observed among the NoV GII samples. Significant genomic signatures of both positive and negative natural selection were identified across the NoV GII genes. A differential pattern of positive selection signals was inferred between the GII.4 and non-GII.4 genotypes. The demographic analysis revealed an increase in the effective population size of NoV GII during 2009-2010, followed by a rapid fall in 2015.
Keywords: genomic diversity; norovirus genogroup II; selection pressure; spatiotemporal.
© 2022. The Author(s), under exclusive licence to Springer-Verlag GmbH Austria, part of Springer Nature.
Conflict of interest statement
The authors declare that they have no competing interests.
Figures

Similar articles
-
GII.4 Norovirus Protease Shows pH-Sensitive Proteolysis with a Unique Arg-His Pairing in the Catalytic Site.J Virol. 2019 Mar 5;93(6):e01479-18. doi: 10.1128/JVI.01479-18. Print 2019 Mar 15. J Virol. 2019. PMID: 30626675 Free PMC article.
-
Genetic diversity and epidemiology of Genogroup II noroviruses in children with acute sporadic gastroenteritis in Shanghai, China, 2012-2017.BMC Infect Dis. 2019 Aug 22;19(1):736. doi: 10.1186/s12879-019-4360-1. BMC Infect Dis. 2019. PMID: 31438883 Free PMC article.
-
Genotyping of noroviruses from patients of the Pilsen University Hospital in the Czech Republic, 2017-2020.Epidemiol Mikrobiol Imunol. 2021 Winter;70(4):233-240. Epidemiol Mikrobiol Imunol. 2021. PMID: 35073701 English.
-
Molecular epidemiology of noroviruses associated with sporadic gastroenteritis in children in Novosibirsk, Russia, 2003-2012.J Med Virol. 2015 May;87(5):740-53. doi: 10.1002/jmv.24068. Epub 2015 Feb 18. J Med Virol. 2015. PMID: 25693507
-
Molecular epidemiology and genotype distributions of noroviruses and sapoviruses in Thailand 2000-2016: A review.J Med Virol. 2018 Apr;90(4):617-624. doi: 10.1002/jmv.25019. Epub 2018 Jan 23. J Med Virol. 2018. PMID: 29315631 Review.
Cited by
-
Elucidating the Implications of Norovirus N- and O-Glycosylation, O-GlcNAcylation, and Phosphorylation.Viruses. 2023 Mar 21;15(3):798. doi: 10.3390/v15030798. Viruses. 2023. PMID: 36992506 Free PMC article. Review.
-
Population genetic analyses unveiled genetic stratification and differential natural selection signatures across the G-gene of viral hemorrhagic septicemia virus.Front Genet. 2022 Dec 12;13:982527. doi: 10.3389/fgene.2022.982527. eCollection 2022. Front Genet. 2022. PMID: 36579328 Free PMC article.
-
Advances in human norovirus research: Vaccines, genotype distribution and antiviral strategies.Virus Res. 2024 Dec;350:199486. doi: 10.1016/j.virusres.2024.199486. Epub 2024 Oct 23. Virus Res. 2024. PMID: 39428038 Free PMC article. Review.
References
-
- Havelaar AH, Kirk MD, Torgerson PR, Gibb HJ, Hald T, Lake RJ, Praet N, Bellinger DC, De Silva NR, Gargouri N, Speybroeck N. World Health Organization global estimates and regional comparisons of the burden of foodborne disease in 2010. PLoS Med. 2015;12(12):e1001923. doi: 10.1371/journal.pmed.1001923. - DOI - PMC - PubMed
MeSH terms
LinkOut - more resources
Full Text Sources
Medical
