

Whole Exome/Genome Sequencing Joint Analysis of a Family with Oligogenic Familial Hypercholesterolemia
- PMID: 35323704
- PMCID: PMC8955453
- DOI: 10.3390/metabo12030262
Whole Exome/Genome Sequencing Joint Analysis of a Family with Oligogenic Familial Hypercholesterolemia
Abstract
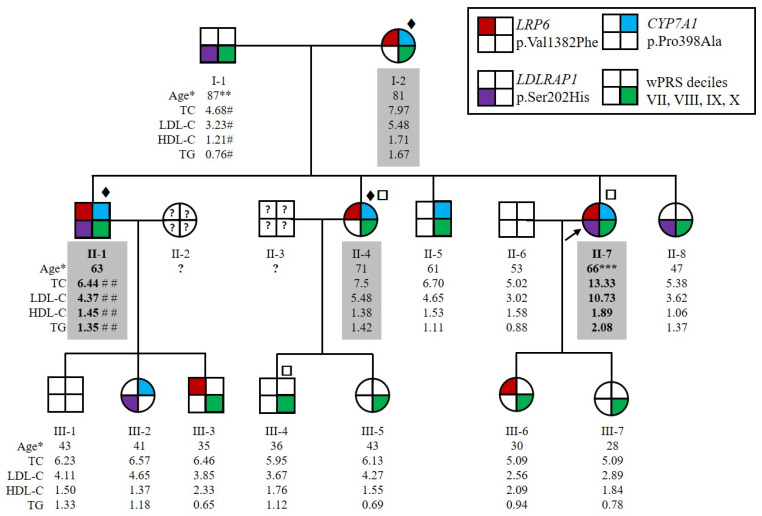
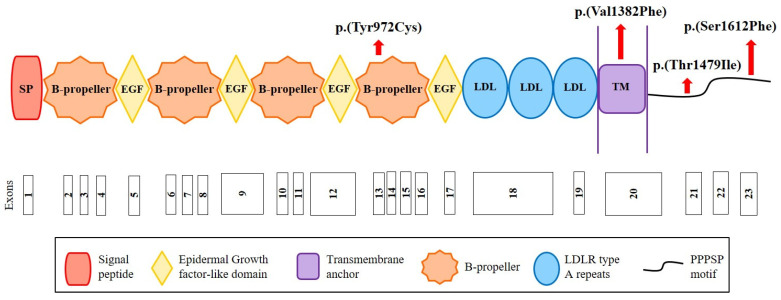
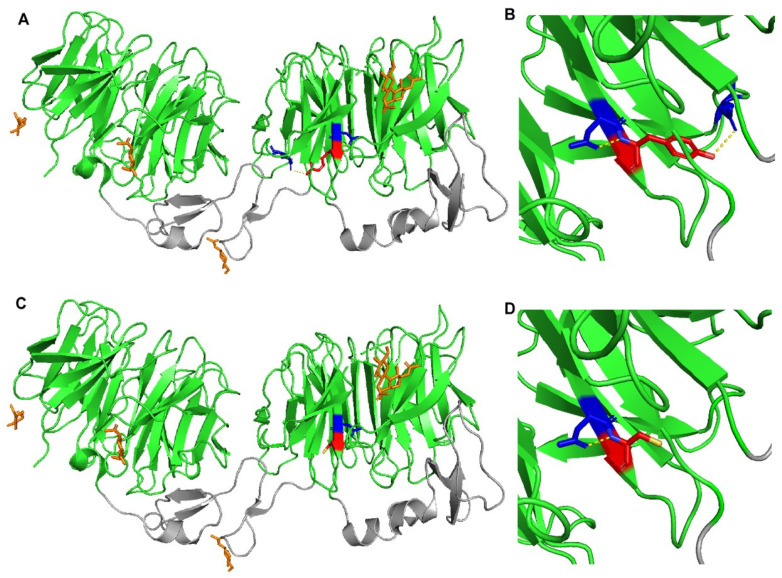
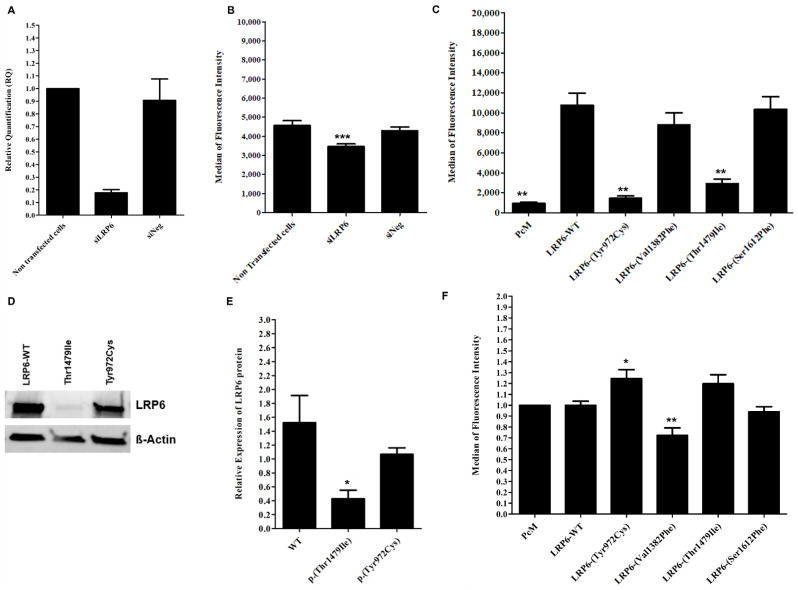
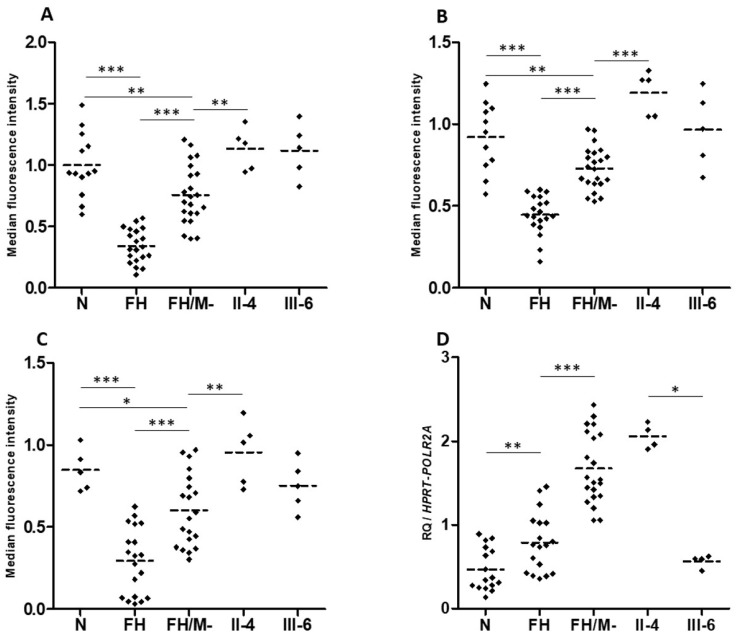
Autosomal Dominant Hypercholesterolemia (ADH) is a genetic disorder caused by pathogenic variants in LDLR, APOB, PCSK9 and APOE genes. We sought to identify new candidate genes responsible for the ADH phenotype in patients without pathogenic variants in the known ADH-causing genes by focusing on a French family with affected and non-affected members who presented a high ADH polygenic risk score (wPRS). Linkage analysis, whole exome and whole genome sequencing resulted in the identification of variants p.(Pro398Ala) in CYP7A1, p.(Val1382Phe) in LRP6 and p.(Ser202His) in LDLRAP1. A total of 6 other variants were identified in 6 of 160 unrelated ADH probands: p.(Ala13Val) and p.(Aps347Asn) in CYP7A1; p.(Tyr972Cys), p.(Thr1479Ile) and p.(Ser1612Phe) in LRP6; and p.(Ser202LeufsTer19) in LDLRAP1. All six probands presented a moderate wPRS. Serum analyses of carriers of the p.(Pro398Ala) variant in CYP7A1 showed no differences in the synthesis of bile acids compared to the serums of non-carriers. Functional studies of the four LRP6 mutants in HEK293T cells resulted in contradictory results excluding a major effect of each variant alone. Within the family, none of the heterozygous for only the LDLRAP1 p.(Ser202His) variant presented ADH. Altogether, each variant individually does not result in elevated LDL-C; however, the oligogenic combination of two or three variants reveals the ADH phenotype.
Keywords: CYP7A1; LDL uptake; LDLRAP1; LRP6; autosomal dominant hypercholesterolemia; linkage analysis; next-generation sequencing; oligogenic hypercholesterolemia; polygenic risk score; protein structural models.
Conflict of interest statement
The authors declare no conflict of interest related with the topic of the paper.
Figures
References
-
- Nordestgaard B.G., Chapman M.J., Humphries S.E., Ginsberg H.N., Masana L., Descamps O.S., Wiklund O., Hegele R.A., Raal F.J., Defesche J.C., et al. Familial hypercholesterolaemia is underdiagnosed and undertreated in the general population: Guidance for clinicians to prevent coronary heart disease: Consensus statement of the European Atherosclerosis Society. Eur. Heart J. 2013;34:3478–3490. doi: 10.1093/eurheartj/eht273. - DOI - PMC - PubMed
-
- Béliard S., Boccara F., Cariou B., Carrié A., Collet X., Farnier M., Ferrières J., Krempf M., Peretti N., Rabès J.-P., et al. High burden of recurrent cardiovascular events in heterozygous familial hypercholesterolemia: The French Familial Hypercholesterolemia Registry. Atherosclerosis. 2018;277:334–340. doi: 10.1016/j.atherosclerosis.2018.08.010. - DOI - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
