

MEN4, the MEN1 Mimicker: A Case Series of three Phenotypically Heterogenous Patients With Unique CDKN1B Mutations
- PMID: 35323929
- PMCID: PMC9282358
- DOI: 10.1210/clinem/dgac162
MEN4, the MEN1 Mimicker: A Case Series of three Phenotypically Heterogenous Patients With Unique CDKN1B Mutations
Abstract
Context: Germline CDKN1B pathogenic variants result in multiple endocrine neoplasia type 4 (MEN4), an autosomal dominant hereditary tumor syndrome variably associated with primary hyperparathyroidism, pituitary adenoma, and duodenopancreatic neuroendocrine tumors.
Objective: To report the phenotype of 3 unrelated cases each with a unique germline CDKN1B variant (of which 2 are novel) and compare these cases with those described in the current literature.
Design/methods: Three case studies, including clinical presentation, germline, and tumor genetic analysis and family history.
Setting: Two tertiary University Hospitals in Sydney, New South Wales, and 1 tertiary University Hospital in Canberra, Australian Capital Territory, Australia.
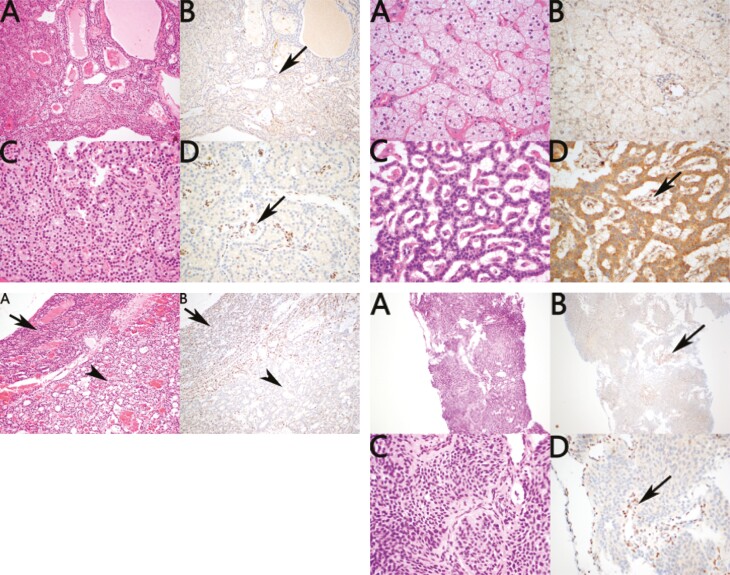
Outcome: Phenotype of the 3 cases and their kindred; molecular analysis and tumor p27kip1 immunohistochemistry.
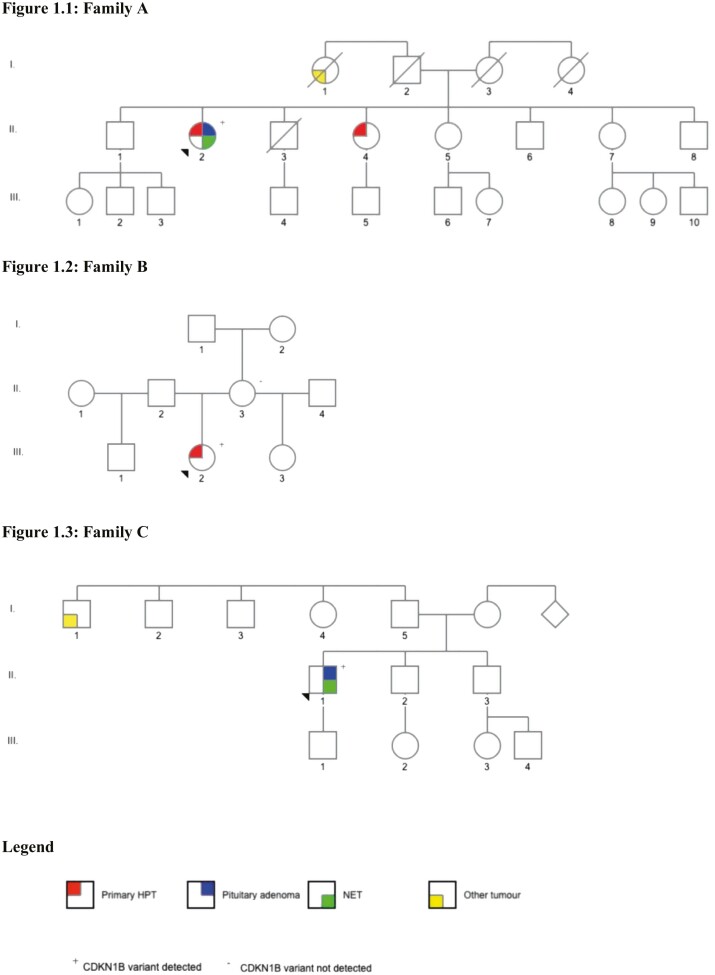
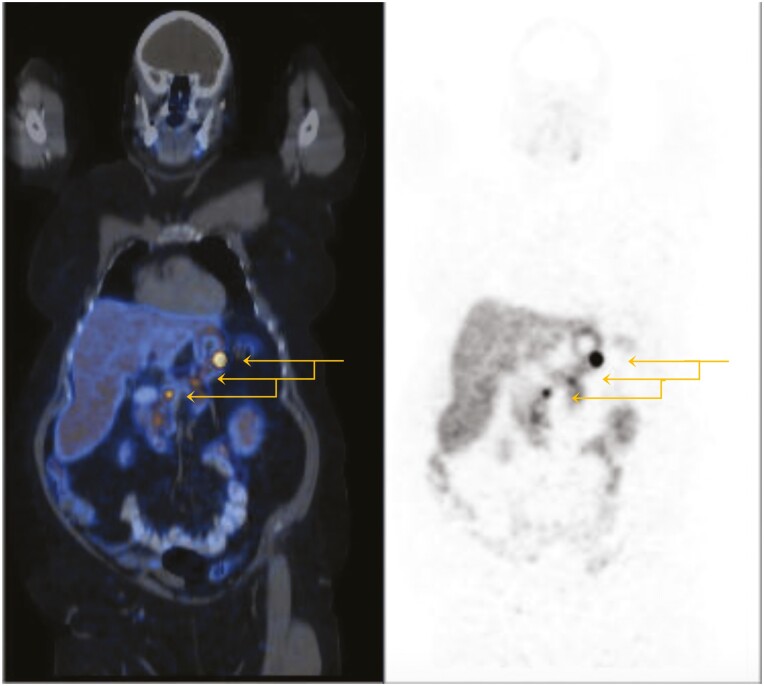
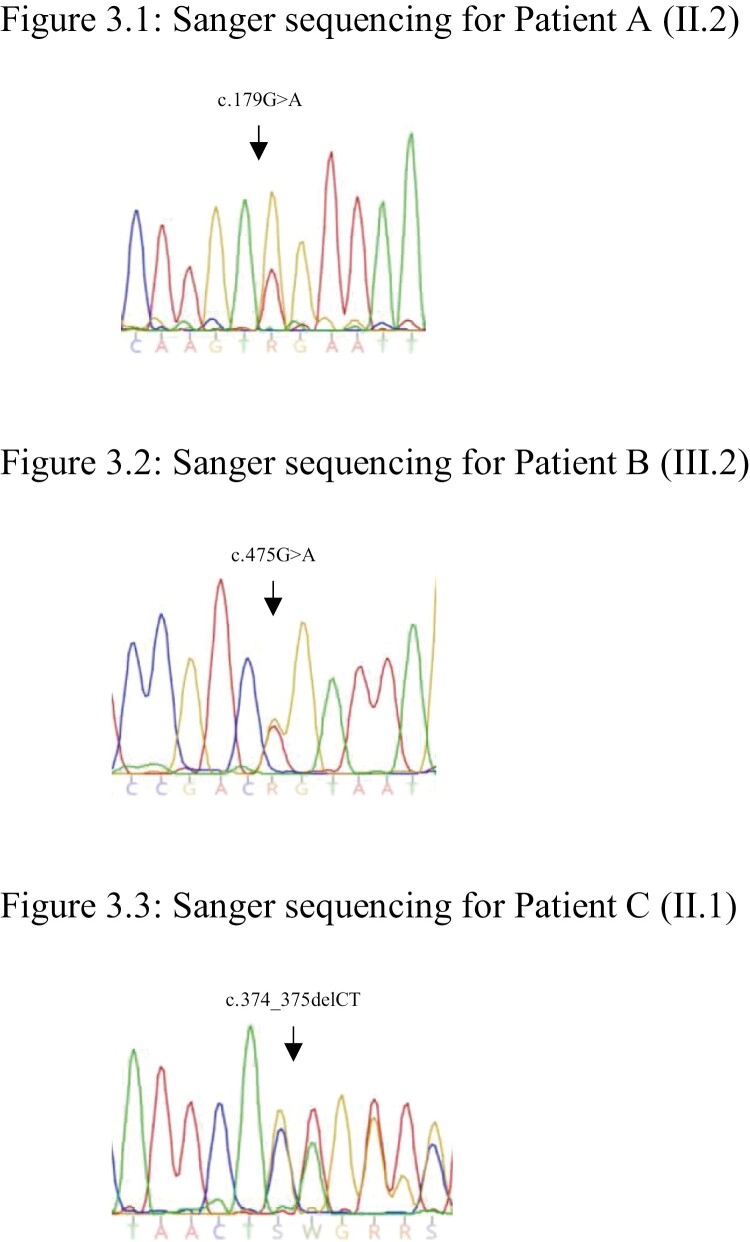
Results: Family A: The proband developed multiglandular primary hyperparathyroidism, a microprolactinoma and a multifocal nonfunctioning duodenopancreatic neuroendocrine tumor. Family B: The proband was diagnosed with primary hyperparathyroidism from a single parathyroid adenoma. Family C: The proband was diagnosed with a nonfunctioning pituitary microadenoma and ectopic Cushing's syndrome from an atypical thymic carcinoid tumor. Germline sequencing in each patient identified a unique variant in CDKN1B, 2 of which are novel (c.179G > A, p.Trp60*; c.475G > A, p.Asp159Asn) and 1 previously reported (c.374_375delCT, p.Ser125*).
Conclusions: Germline CDKN1B pathogenic variants cause the syndrome MEN4. The phenotype resulting from the 3 pathogenic variants described in this series highlights the heterogenous nature of this syndrome, ranging from isolated primary hyperparathyroidism to the full spectrum of endocrine manifestations. We report the first described cases of a prolactinoma and an atypical thymic carcinoid tumor in MEN4.
Keywords: CDKN1B germline mutation; MEN4; atypical carcinoid tumor; multiple endocrine neoplasia type 4; pancreatic neuroendocrine tumor; pituitary adenoma; primary hyperparathyroidism.
© The Author(s) 2022. Published by Oxford University Press on behalf of the Endocrine Society.
Figures
Similar articles
-
Clinical Features of Multiple Endocrine Neoplasia Type 4: Novel Pathogenic Variant and Review of Published Cases.J Clin Endocrinol Metab. 2019 Sep 1;104(9):3637-3646. doi: 10.1210/jc.2019-00082. J Clin Endocrinol Metab. 2019. PMID: 30990521 Free PMC article. Review.
-
A heterozygous frameshift mutation in exon 1 of CDKN1B gene in a patient affected by MEN4 syndrome.Eur J Endocrinol. 2014 Aug;171(2):K7-K17. doi: 10.1530/EJE-14-0080. Epub 2014 May 12. Eur J Endocrinol. 2014. PMID: 24819502
-
Beyond MEN1, When to Think About MEN4? Retrospective Study on 5600 Patients in the French Population and Literature Review.J Clin Endocrinol Metab. 2024 Jun 17;109(7):e1482-e1493. doi: 10.1210/clinem/dgae055. J Clin Endocrinol Metab. 2024. PMID: 38288531 Review.
-
Germline CDKN1B variant type and site are associated with phenotype in MEN4.Endocr Relat Cancer. 2022 Dec 12;30(1):e220174. doi: 10.1530/ERC-22-0174. Print 2023 Jan 1. Endocr Relat Cancer. 2022. PMID: 36256846 Review.
-
Novel germline variants of CDKN1B and CDKN2C identified during screening for familial primary hyperparathyroidism.J Endocrinol Invest. 2023 Apr;46(4):829-840. doi: 10.1007/s40618-022-01948-7. Epub 2022 Nov 5. J Endocrinol Invest. 2023. PMID: 36334246 Free PMC article.
Cited by
-
Syndromic MEN1 parathyroid adenomas consist of both subclonal nodules and clonally independent tumors.Virchows Arch. 2024 May;484(5):789-798. doi: 10.1007/s00428-023-03730-3. Epub 2024 Jan 20. Virchows Arch. 2024. PMID: 38244045 Free PMC article.
-
[Special features of the diagnostics and treatment of hereditary primary hyperparathyroidism].Chirurgie (Heidelb). 2023 Jul;94(7):586-594. doi: 10.1007/s00104-023-01897-8. Epub 2023 Jun 8. Chirurgie (Heidelb). 2023. PMID: 37291366 Review. German.
-
Multiple endocrine neoplasia type 4: a new member of the MEN family.Endocr Connect. 2023 Jan 24;12(2):e220411. doi: 10.1530/EC-22-0411. Print 2023 Feb 1. Endocr Connect. 2023. PMID: 36520683 Free PMC article.
-
Turning Points in Cross-Disciplinary Perspective of Primary Hyperparathyroidism and Pancreas Involvements: Hypercalcemia-Induced Pancreatitis, MEN1 Gene-Related Tumors, and Insulin Resistance.Int J Mol Sci. 2024 Jun 8;25(12):6349. doi: 10.3390/ijms25126349. Int J Mol Sci. 2024. PMID: 38928056 Free PMC article. Review.
-
A Pituitary Macroadenoma Cosecreting Prolactin and Growth Hormone in a Patient With Multiple Endocrine Neoplasia Type 4.JCEM Case Rep. 2025 May 29;3(7):luaf124. doi: 10.1210/jcemcr/luaf124. eCollection 2025 Jul. JCEM Case Rep. 2025. PMID: 40443455 Free PMC article.
References
-
- Romanet P, Mohamed A, Giraud S, et al. . UMD-MEN1 database: an overview of the 370 MEN1 variants present in 1676 patients from the French population. J Clin Endocrinol Metab. 2019;104(3):753-764. - PubMed
-
- Kooblall KG, Boon H, Cranston T, et al. . Multiple endocrine neoplasia type 1 (MEN1) 5′UTR deletion, in MEN1 family, decreases Menin expression. J Bone Miner Res. 2021; 36(1):100-109. - PubMed
-
- Burgess JR, Nord B, David R, et al. . Phenotype and phenocopy: the relationship between genotype and clinical phenotype in a single large family with multiple endocrine neoplasia type 1 (MEN 1). Clin Endocrinol (Oxf). 2000;53(2):205-211. - PubMed
-
- Turner JJ, Christie PT, Pearce SH, Turnpenny PD, Thakker RV. Diagnostic challenges due to phenocopies: lessons from multiple endocrine neoplasia type1 (MEN1). Hum Mutat. 2010;31(1):E1089-E1101. - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
