

Integrating Long-Range Regulatory Interactions to Predict Gene Expression Using Graph Convolutional Networks
- PMID: 35325548
- PMCID: PMC9125570
- DOI: 10.1089/cmb.2021.0316
Integrating Long-Range Regulatory Interactions to Predict Gene Expression Using Graph Convolutional Networks
Abstract
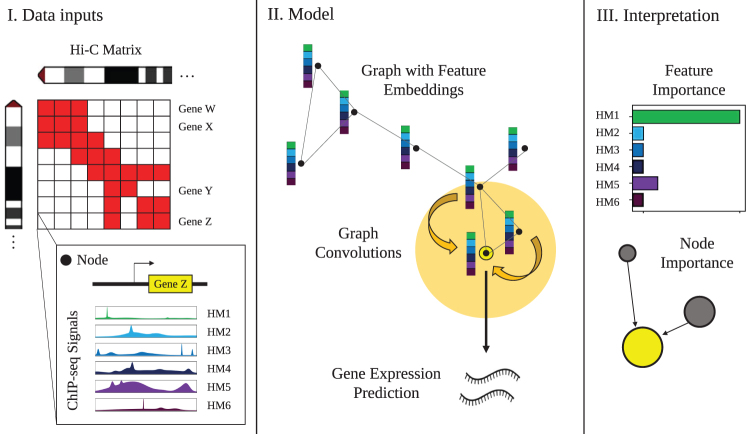
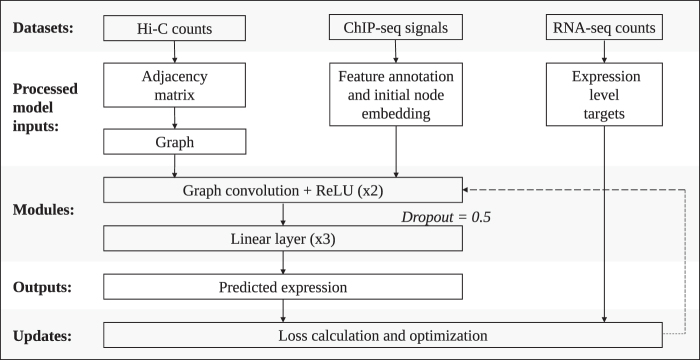
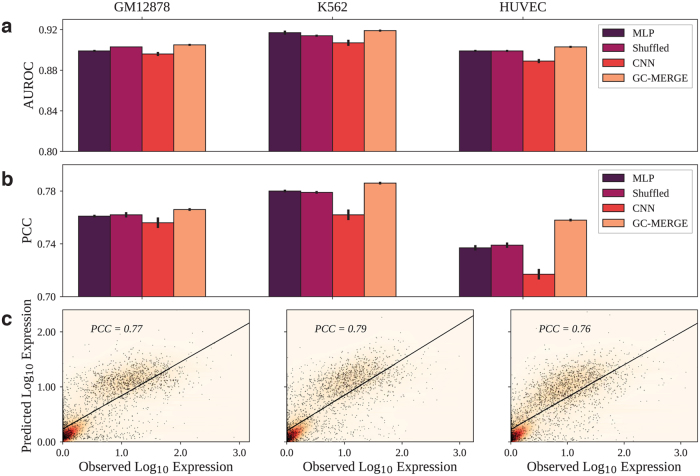
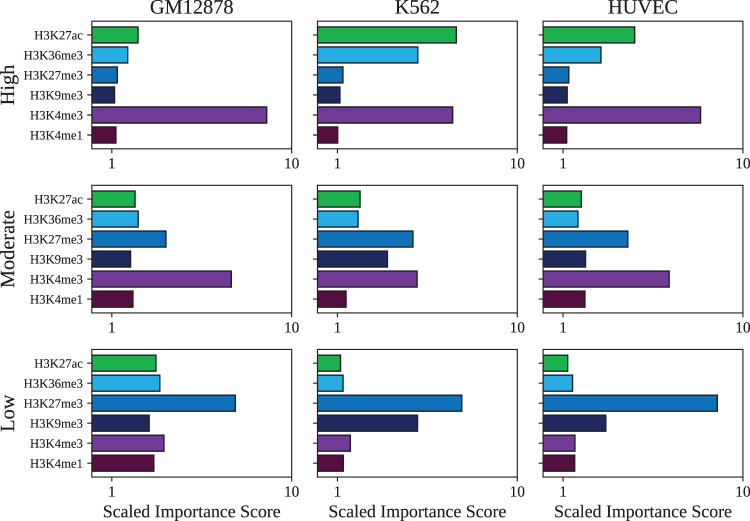
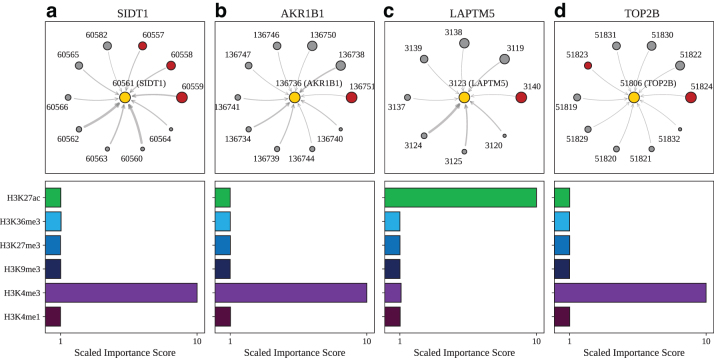
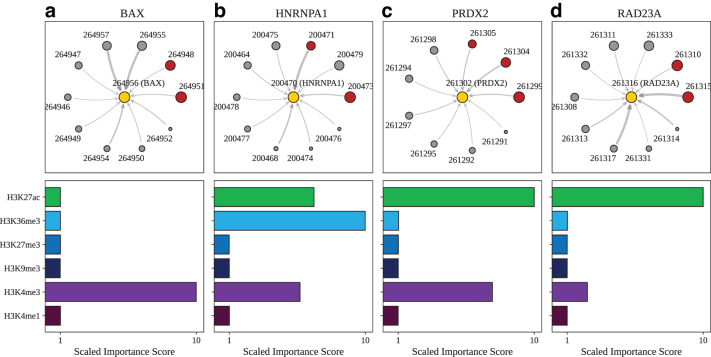
Long-range regulatory interactions among genomic regions are critical for controlling gene expression, and their disruption has been associated with a host of diseases. However, when modeling the effects of regulatory factors, most deep learning models either neglect long-range interactions or fail to capture the inherent 3D structure of the underlying genomic organization. To address these limitations, we present a Graph Convolutional Model for Epigenetic Regulation of Gene Expression (GC-MERGE). Using a graph-based framework, the model incorporates important information about long-range interactions via a natural encoding of genomic spatial interactions into the graph representation. It integrates measurements of both the global genomic organization and the local regulatory factors, specifically histone modifications, to not only predict the expression of a given gene of interest but also quantify the importance of its regulatory factors. We apply GC-MERGE to data sets for three cell lines-GM12878 (lymphoblastoid), K562 (myelogenous leukemia), and HUVEC (human umbilical vein endothelial)-and demonstrate its state-of-the-art predictive performance. Crucially, we show that our model is interpretable in terms of the observed biological regulatory factors, highlighting both the histone modifications and the interacting genomic regions contributing to a gene's predicted expression. We provide model explanations for multiple exemplar genes and validate them with evidence from the literature. Our model presents a novel setup for predicting gene expression by integrating multimodal data sets in a graph convolutional framework. More importantly, it enables interpretation of the biological mechanisms driving the model's predictions.
Keywords: Hi-C; deep learning; gene expression; graph neural networks; histone modifications.
Conflict of interest statement
The authors declare they have no conflicting financial interests.
Figures
References
-
- Agarwal, V., and Shendure, J.. 2020. Predicting mRNA abundance directly from genomic sequence using deep convolutional neural networks. Cell Rep. 31, 107663. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
