

Important Functions and Molecular Mechanisms of Mitochondrial Redox Signaling in Pulmonary Hypertension
- PMID: 35326123
- PMCID: PMC8944689
- DOI: 10.3390/antiox11030473
Important Functions and Molecular Mechanisms of Mitochondrial Redox Signaling in Pulmonary Hypertension
Abstract
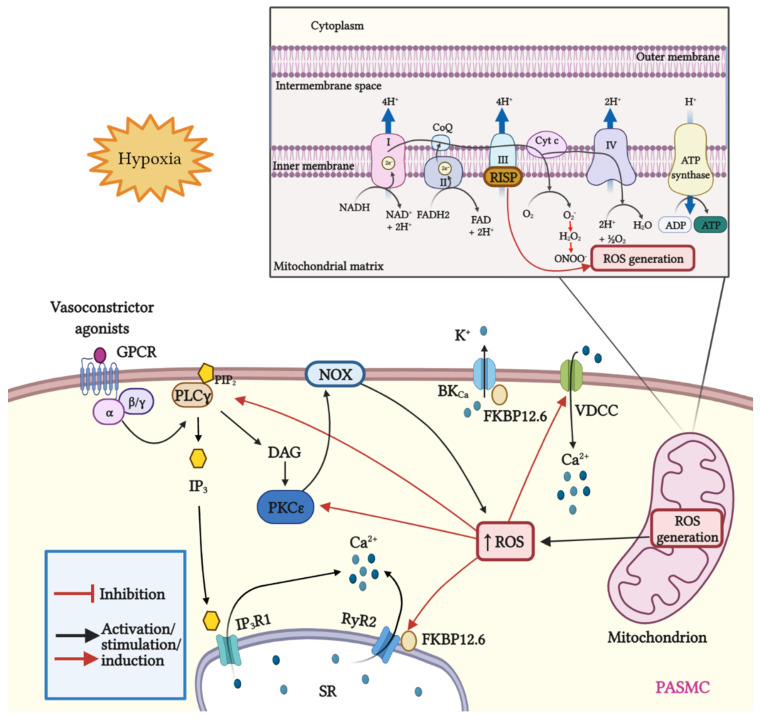
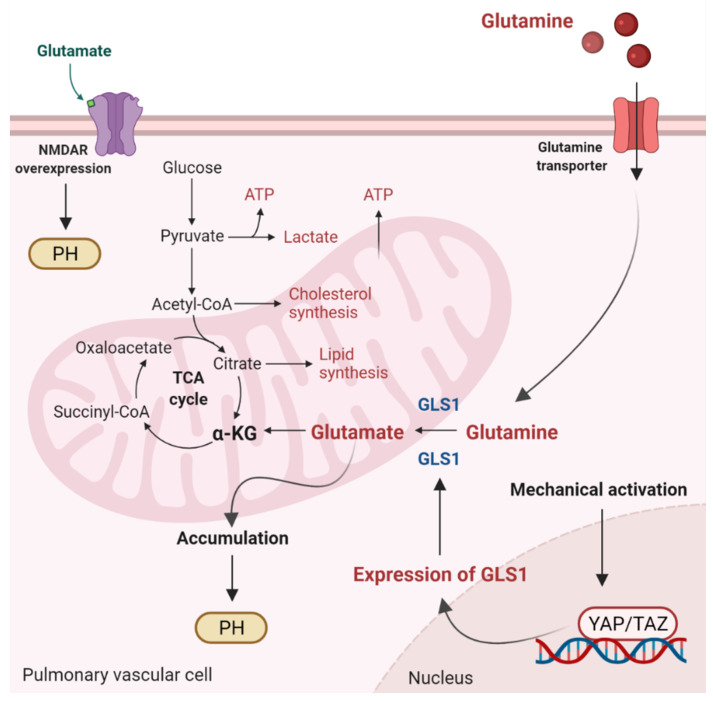
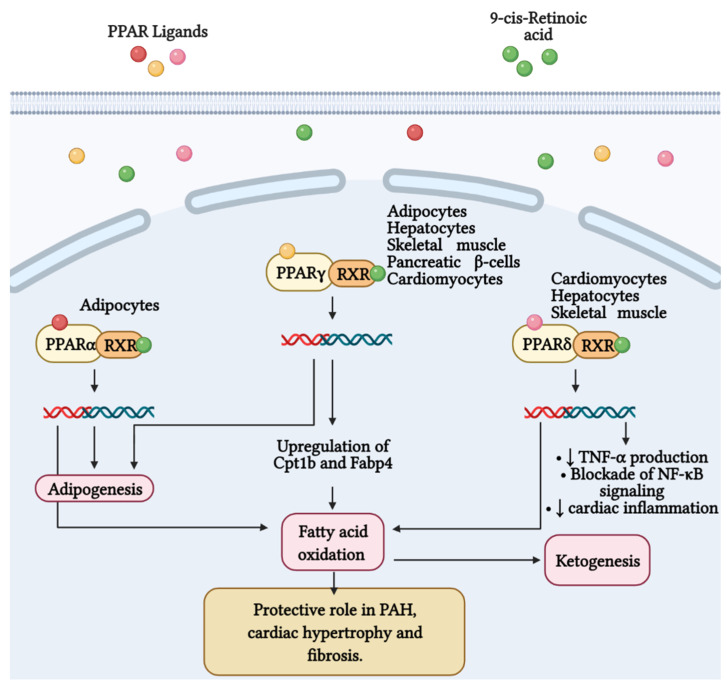
Mitochondria are important organelles that act as a primary site to produce reactive oxygen species (ROS). Additionally, mitochondria play a pivotal role in the regulation of Ca2+ signaling, fatty acid oxidation, and ketone synthesis. Dysfunction of these signaling molecules leads to the development of pulmonary hypertension (PH), atherosclerosis, and other vascular diseases. Features of PH include vasoconstriction and pulmonary artery (PA) remodeling, which can result from abnormal proliferation, apoptosis, and migration of PA smooth muscle cells (PASMCs). These responses are mediated by increased Rieske iron-sulfur protein (RISP)-dependent mitochondrial ROS production and increased mitochondrial Ca2+ levels. Mitochondrial ROS and Ca2+ can both synergistically activate nuclear factor κB (NF-κB) to trigger inflammatory responses leading to PH, right ventricular failure, and death. Evidence suggests that increased mitochondrial ROS and Ca2+ signaling leads to abnormal synthesis of ketones, which play a critical role in the development of PH. In this review, we discuss some of the recent findings on the important interactive role and molecular mechanisms of mitochondrial ROS and Ca2+ in the development and progression of PH. We also address the contributions of NF-κB-dependent inflammatory responses and ketone-mediated oxidative stress due to abnormal regulation of mitochondrial ROS and Ca2+ signaling in PH.
Keywords: Ca2+ signaling; ketones; mitochondrial ROS; pulmonary hypertension.
Conflict of interest statement
The authors declare no conflict of interests. The funders had no role in the design of the study, analyses, or interpretation or in the writing of the manuscript. Figures in this manuscript were created with BioRender.com.
Figures
Similar articles
-
Cross Talk Between Mitochondrial Reactive Oxygen Species and Sarcoplasmic Reticulum Calcium in Pulmonary Arterial Smooth Muscle Cells.Adv Exp Med Biol. 2017;967:289-298. doi: 10.1007/978-3-319-63245-2_17. Adv Exp Med Biol. 2017. PMID: 29047093 Review.
-
Important Role of Sarcoplasmic Reticulum Ca2+ Release via Ryanodine Receptor-2 Channel in Hypoxia-Induced Rieske Iron-Sulfur Protein-Mediated Mitochondrial Reactive Oxygen Species Generation in Pulmonary Artery Smooth Muscle Cells.Antioxid Redox Signal. 2020 Mar 1;32(7):447-462. doi: 10.1089/ars.2018.7652. Epub 2019 Oct 11. Antioxid Redox Signal. 2020. PMID: 31456413 Free PMC article.
-
Endoplasmic reticulum Ca2+ release causes Rieske iron-sulfur protein-mediated mitochondrial ROS generation in pulmonary artery smooth muscle cells.Biosci Rep. 2019 Dec 20;39(12):BSR20192414. doi: 10.1042/BSR20192414. Biosci Rep. 2019. Retraction in: Biosci Rep. 2020 Apr 30;40(4):BSR-20192414_RET. doi: 10.1042/BSR-20192414_RET. PMID: 31710081 Free PMC article. Retracted.
-
Mitochondrial Rieske iron-sulfur protein in pulmonary artery smooth muscle: A key primary signaling molecule in pulmonary hypertension.Arch Biochem Biophys. 2020 Apr 15;683:108234. doi: 10.1016/j.abb.2019.108234. Epub 2020 Jan 21. Arch Biochem Biophys. 2020. PMID: 31980131
-
Mitochondrial Rieske iron-sulfur protein in pulmonary artery smooth muscle: A key primary signaling molecule in pulmonary hypertension.Arch Biochem Biophys. 2019 Mar 30;664:68-75. doi: 10.1016/j.abb.2019.01.029. Epub 2019 Jan 30. Arch Biochem Biophys. 2019. PMID: 30710505 Review.
Cited by
-
The Potential Important Role of Mitochondrial Rieske Iron-Sulfur Protein as a Novel Therapeutic Target for Pulmonary Hypertension in Chronic Obstructive Pulmonary Disease.Biomedicines. 2022 Apr 21;10(5):957. doi: 10.3390/biomedicines10050957. Biomedicines. 2022. PMID: 35625694 Free PMC article. Review.
-
Salidroside enhances NO bioavailability and modulates arginine metabolism to alleviate pulmonary arterial hypertension.Eur J Med Res. 2024 Aug 17;29(1):423. doi: 10.1186/s40001-024-02016-x. Eur J Med Res. 2024. PMID: 39152472 Free PMC article.
-
In Vitro Cell Death Mechanisms Induced by Dicoma anomala Root Extract in Combination with ZnPcS4 Mediated-Photodynamic Therapy in A549 Lung Cancer Cells.Cells. 2022 Oct 19;11(20):3288. doi: 10.3390/cells11203288. Cells. 2022. PMID: 36291155 Free PMC article.
-
Hypoxia-Induced Mitochondrial ROS and Function in Pulmonary Arterial Endothelial Cells.Cells. 2024 Nov 1;13(21):1807. doi: 10.3390/cells13211807. Cells. 2024. PMID: 39513914 Free PMC article.
-
Biomimetic Integrated Nanozyme for Flare and Recurrence of Gouty Arthritis.Asian J Pharm Sci. 2024 Jun;19(3):100913. doi: 10.1016/j.ajps.2024.100913. Epub 2024 Apr 18. Asian J Pharm Sci. 2024. PMID: 38903129 Free PMC article.
References
-
- Waypa G.B., Marks J.D., Guzy R.D., Mungai P.T., Schriewer J.M., Dokic D., Ball M.K., Schumacker P.T. Superoxide generated at mitochondrial complex III triggers acute responses to hypoxia in the pulmonary circulation. Am. J. Respir. Crit. Care Med. 2013;187:424–432. doi: 10.1164/rccm.201207-1294OC. - DOI - PMC - PubMed
-
- Mei L., Zheng Y.-M., Song T., Yadav V.R., Joseph L.C., Truong L., Kandhi S., Barroso M.M., Takeshima H., Judson M.A., et al. Rieske iron-sulfur protein induces FKBP12.6/RyR2 complex remodeling and subsequent pulmonary hypertension through NF-κB/cyclin D1 pathway. Nat. Commun. 2020;11:3527. doi: 10.1038/s41467-020-17314-1. - DOI - PMC - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
