

MicroRNAs, Long Non-Coding RNAs, and Circular RNAs in the Redox Control of Cell Senescence
- PMID: 35326131
- PMCID: PMC8944605
- DOI: 10.3390/antiox11030480
MicroRNAs, Long Non-Coding RNAs, and Circular RNAs in the Redox Control of Cell Senescence
Abstract
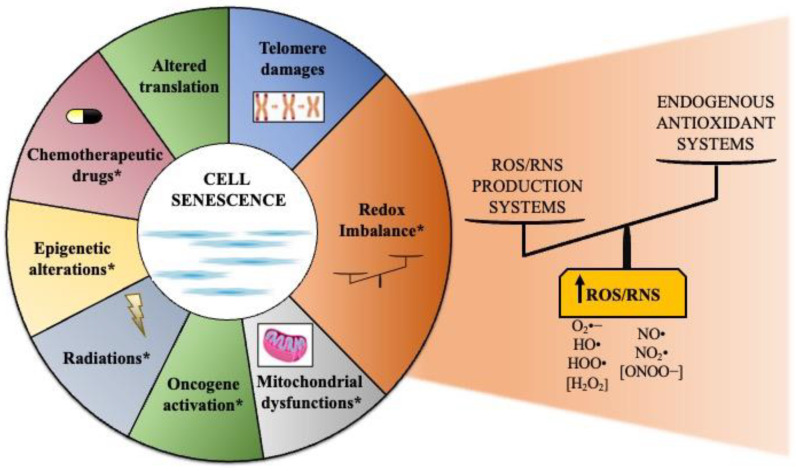
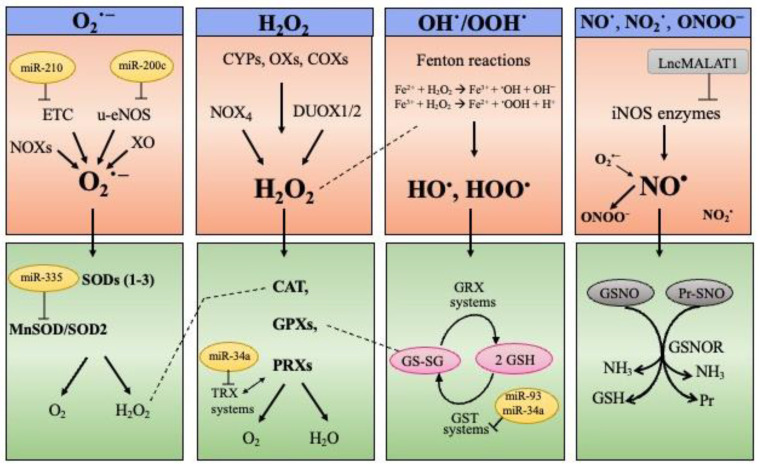
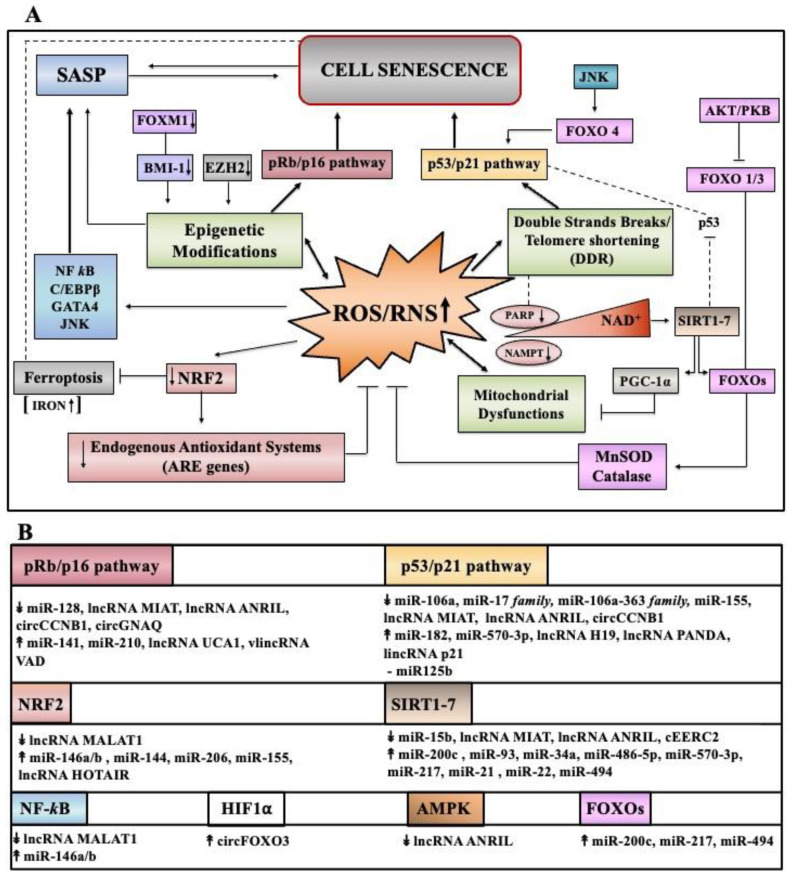
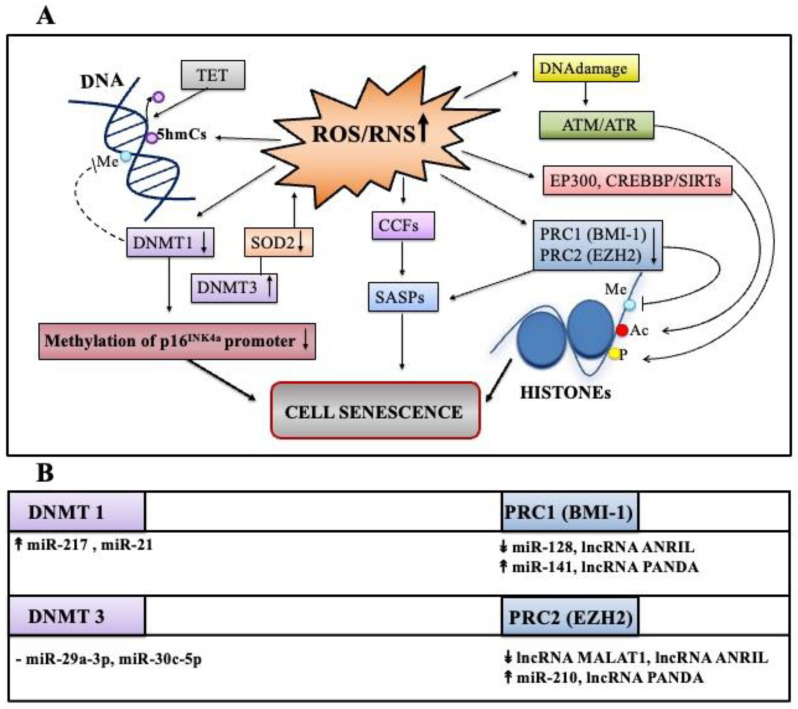
Cell senescence is critical in diverse aspects of organism life. It is involved in tissue development and homeostasis, as well as in tumor suppression. Consequently, it is tightly integrated with basic physiological processes during life. On the other hand, senescence is gradually being considered as a major contributor of organismal aging and age-related diseases. Increased oxidative stress is one of the main risk factors for cellular damages, and thus a driver of senescence. In fact, there is an intimate link between cell senescence and response to different types of cellular stress. Oxidative stress occurs when the production of reactive oxygen species/reactive nitrogen species (ROS/RNS) is not adequately detoxified by the antioxidant defense systems. Non-coding RNAs are endogenous transcripts that govern gene regulatory networks, thus impacting both physiological and pathological events. Among these molecules, microRNAs, long non-coding RNAs, and more recently circular RNAs are considered crucial mediators of almost all cellular processes, including those implicated in oxidative stress responses. Here, we will describe recent data on the link between ROS/RNS-induced senescence and the current knowledge on the role of non-coding RNAs in the senescence program.
Keywords: aging; cell senescence; circular RNAs; long non-coding RNAs; microRNAs; non-coding RNAs; oxidative stress; redox homeostasis.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
Similar articles
-
Non-coding RNAs as Regulators of Cellular Senescence in Idiopathic Pulmonary Fibrosis and Chronic Obstructive Pulmonary Disease.Front Med (Lausanne). 2020 Dec 23;7:603047. doi: 10.3389/fmed.2020.603047. eCollection 2020. Front Med (Lausanne). 2020. PMID: 33425948 Free PMC article. Review.
-
Emerging Role of Non-Coding RNAs in Senescence.Front Cell Dev Biol. 2022 Jul 5;10:869011. doi: 10.3389/fcell.2022.869011. eCollection 2022. Front Cell Dev Biol. 2022. PMID: 35865636 Free PMC article. Review.
-
The Role of Non-Coding RNAs in the Neuroprotective Effects of Glutathione.Int J Mol Sci. 2021 Apr 19;22(8):4245. doi: 10.3390/ijms22084245. Int J Mol Sci. 2021. PMID: 33921907 Free PMC article. Review.
-
Regulation of cellular senescence by microRNAs.Mech Ageing Dev. 2020 Jul;189:111264. doi: 10.1016/j.mad.2020.111264. Epub 2020 May 23. Mech Ageing Dev. 2020. PMID: 32450085 Review.
-
MicroRNA controls of cellular senescence.BMB Rep. 2018 Oct;51(10):493-499. doi: 10.5483/BMBRep.2018.51.10.209. BMB Rep. 2018. PMID: 30269742 Free PMC article.
Cited by
-
The Role of Antioxidants in the Interplay between Oxidative Stress and Senescence.Antioxidants (Basel). 2022 Jun 22;11(7):1224. doi: 10.3390/antiox11071224. Antioxidants (Basel). 2022. PMID: 35883714 Free PMC article. Review.
-
Fundamentals of redox regulation in biology.Nat Rev Mol Cell Biol. 2024 Sep;25(9):701-719. doi: 10.1038/s41580-024-00730-2. Epub 2024 Apr 30. Nat Rev Mol Cell Biol. 2024. PMID: 38689066 Free PMC article. Review.
-
Chondrocyte Homeostasis and Differentiation: Transcriptional Control and Signaling in Healthy and Osteoarthritic Conditions.Life (Basel). 2023 Jun 28;13(7):1460. doi: 10.3390/life13071460. Life (Basel). 2023. PMID: 37511835 Free PMC article. Review.
-
The Molecular Interplay Between p53-Mediated Ferroptosis and Non-Coding RNAs in Cancer.Int J Mol Sci. 2025 Jul 9;26(14):6588. doi: 10.3390/ijms26146588. Int J Mol Sci. 2025. PMID: 40724837 Free PMC article. Review.
-
The Role of Hydrogen Sulfide (H2S) in Epigenetic Regulation of Neurodegenerative Diseases: A Systematic Review.Int J Mol Sci. 2023 Aug 8;24(16):12555. doi: 10.3390/ijms241612555. Int J Mol Sci. 2023. PMID: 37628735 Free PMC article.
References
-
- Storer M., Mas A., Robert-Moreno A., Pecoraro M., Ortells M.C., di Giacomo V., Yosef R., Pilpel N., Krizhanovsky V., Sharpe J., et al. Senescence is a developmental mechanism that contributes to embryonic growth and patterning. Cell. 2013;155:1119–1130. doi: 10.1016/j.cell.2013.10.041. - DOI - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
