

Chemotherapy Resistance: Role of Mitochondrial and Autophagic Components
- PMID: 35326612
- PMCID: PMC8945922
- DOI: 10.3390/cancers14061462
Chemotherapy Resistance: Role of Mitochondrial and Autophagic Components
Abstract
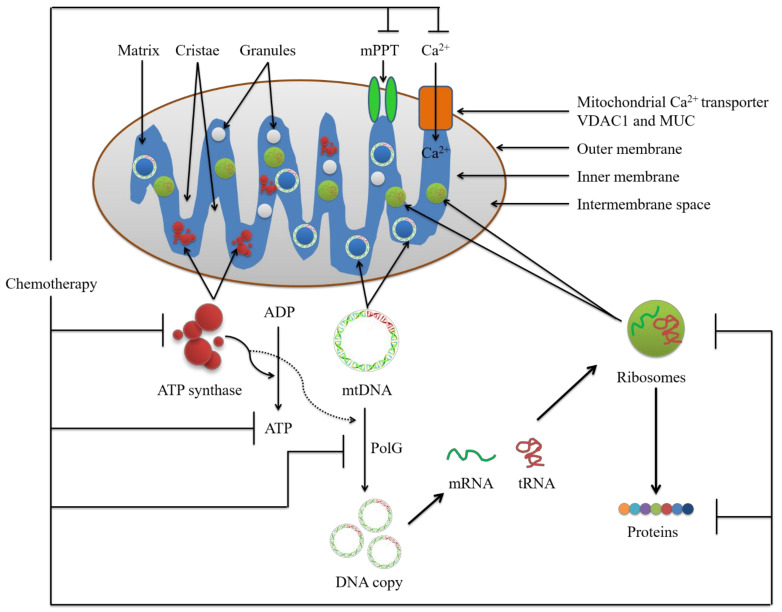
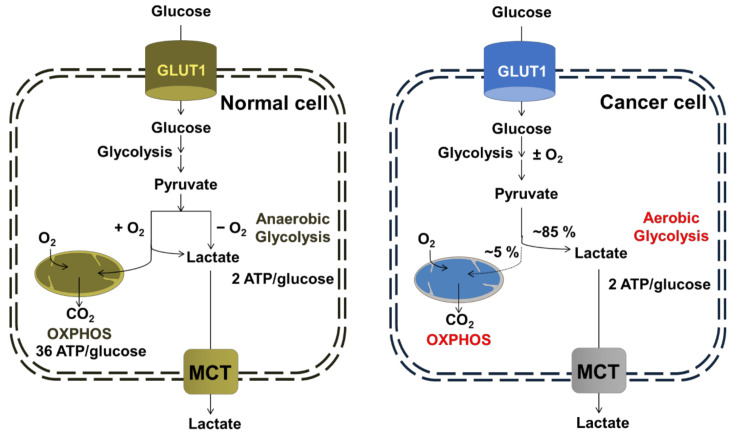
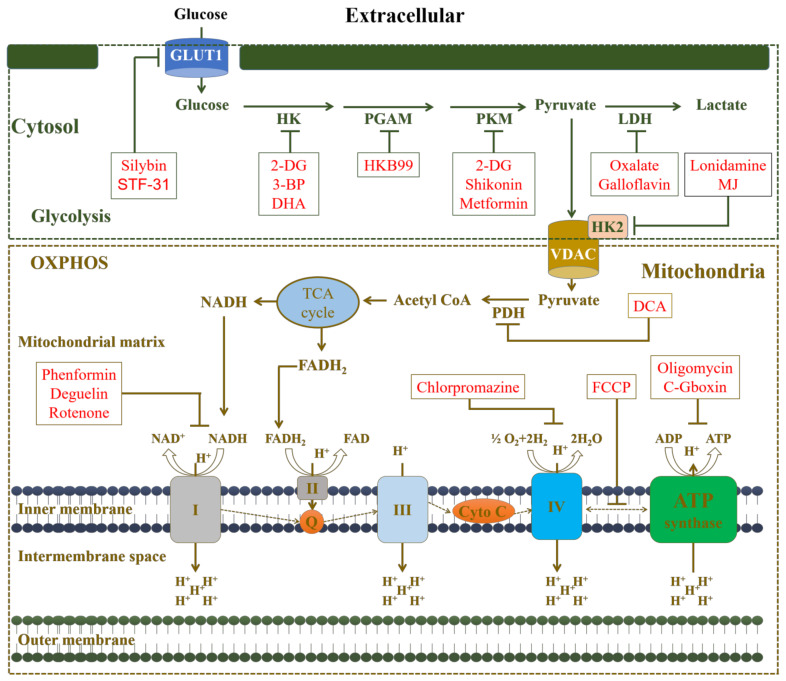
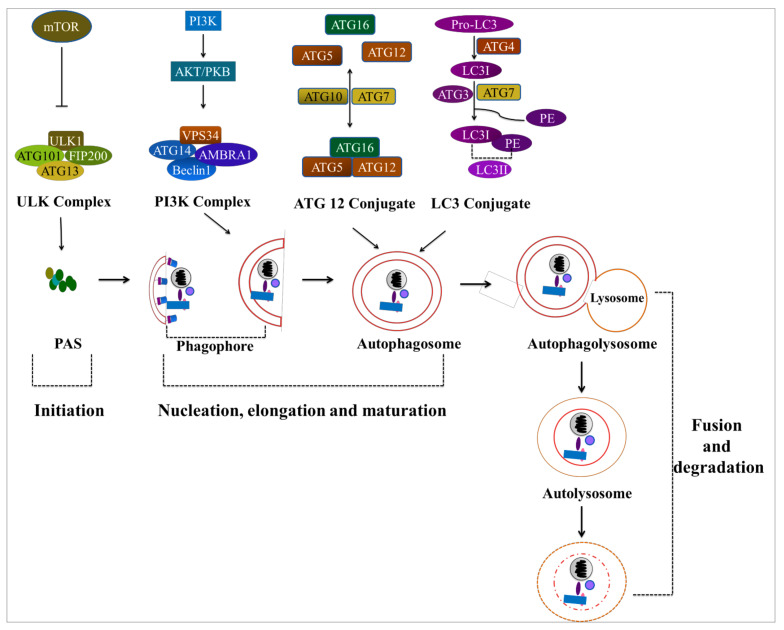
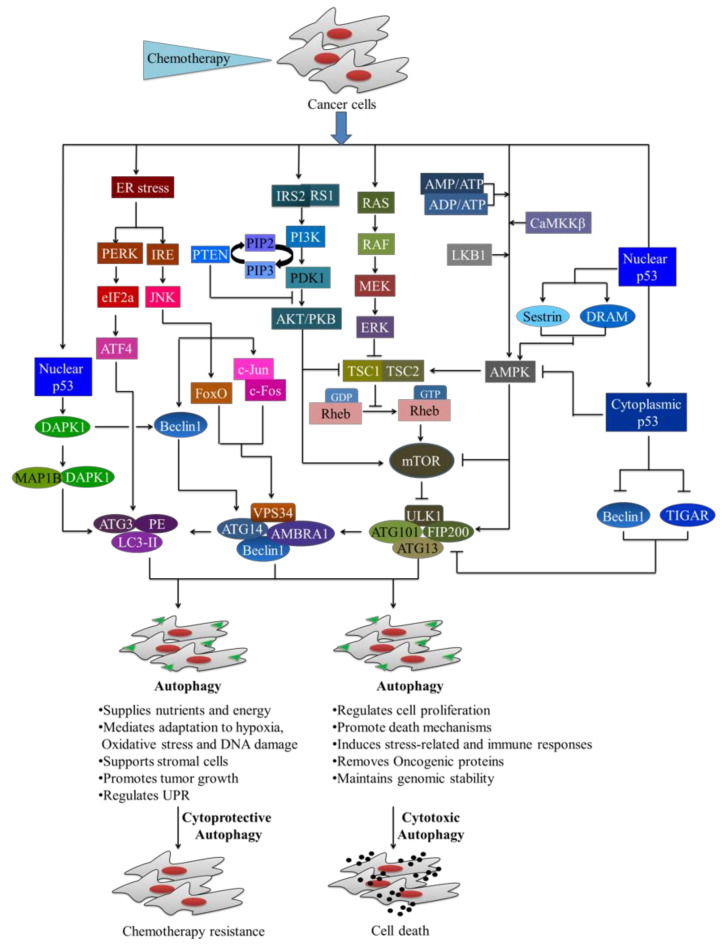
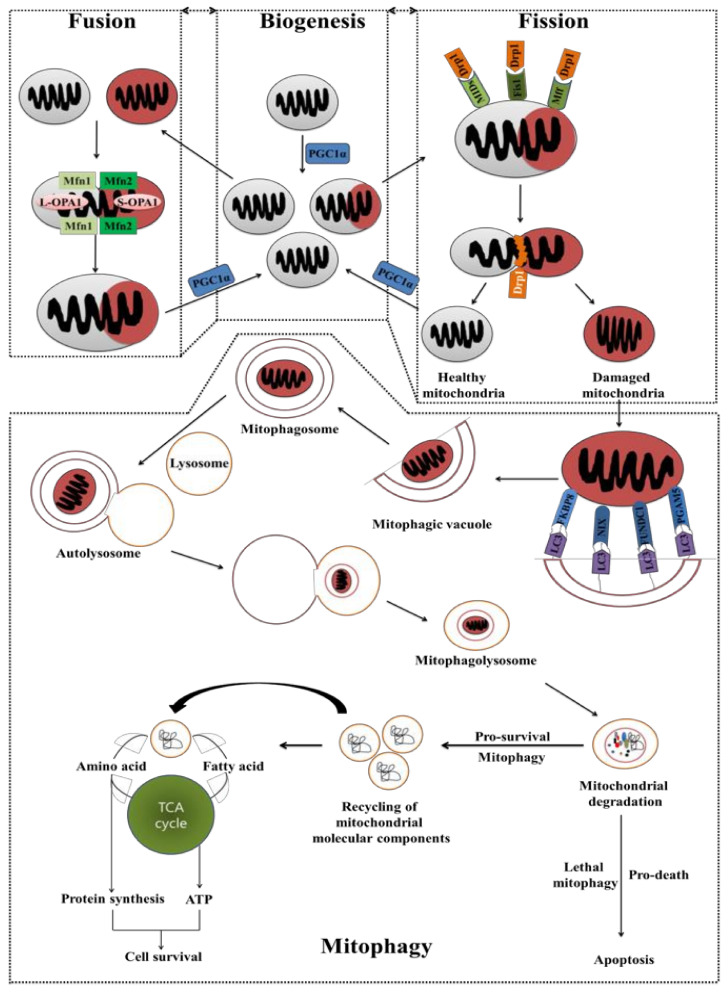
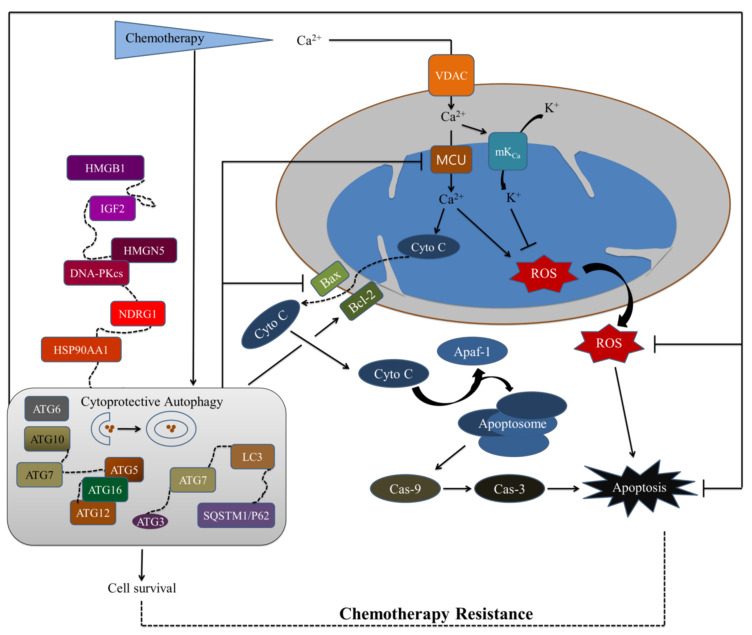
Cancer chemotherapy resistance is one of the most critical obstacles in cancer therapy. One of the well-known mechanisms of chemotherapy resistance is the change in the mitochondrial death pathways which occur when cells are under stressful situations, such as chemotherapy. Mitophagy, or mitochondrial selective autophagy, is critical for cell quality control because it can efficiently break down, remove, and recycle defective or damaged mitochondria. As cancer cells use mitophagy to rapidly sweep away damaged mitochondria in order to mediate their own drug resistance, it influences the efficacy of tumor chemotherapy as well as the degree of drug resistance. Yet despite the importance of mitochondria and mitophagy in chemotherapy resistance, little is known about the precise mechanisms involved. As a consequence, identifying potential therapeutic targets by analyzing the signal pathways that govern mitophagy has become a vital research goal. In this paper, we review recent advances in mitochondrial research, mitophagy control mechanisms, and their implications for our understanding of chemotherapy resistance.
Keywords: chemotherapy resistance; mitochondria; mitophagy.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
Similar articles
-
The Role of Mitochondrial Dynamics and Mitophagy in Carcinogenesis, Metastasis and Therapy.Front Cell Dev Biol. 2020 Jun 10;8:413. doi: 10.3389/fcell.2020.00413. eCollection 2020. Front Cell Dev Biol. 2020. PMID: 32587855 Free PMC article. Review.
-
Mitophagy: Link to cancer development and therapy.Biochem Biophys Res Commun. 2017 Jan 15;482(3):432-439. doi: 10.1016/j.bbrc.2016.10.088. Epub 2017 Feb 3. Biochem Biophys Res Commun. 2017. PMID: 28212727 Review.
-
Mitochondrial rewiring through mitophagy and mitochondrial biogenesis in cancer stem cells: A potential target for anti-CSC cancer therapy.Cancer Lett. 2021 Feb 1;498:217-228. doi: 10.1016/j.canlet.2020.10.036. Epub 2020 Nov 10. Cancer Lett. 2021. PMID: 33186655 Review.
-
Leukemia and mitophagy: a novel perspective for understanding oncogenesis and resistance.Ann Hematol. 2024 Jul;103(7):2185-2196. doi: 10.1007/s00277-024-05635-w. Epub 2024 Jan 29. Ann Hematol. 2024. PMID: 38282059 Review.
-
Mitochondria autophagy: a potential target for cancer therapy.J Drug Target. 2021 Jul;29(6):576-591. doi: 10.1080/1061186X.2020.1867992. Epub 2021 Feb 8. J Drug Target. 2021. PMID: 33554661 Review.
Cited by
-
Root Bark Extract of Oroxylum indicum Vent. Inhibits Solid and Ascites Tumors and Prevents the Development of DMBA-Induced Skin Papilloma Formation.Molecules. 2022 Dec 2;27(23):8459. doi: 10.3390/molecules27238459. Molecules. 2022. PMID: 36500567 Free PMC article.
-
Recent Update and Drug Target in Molecular and Pharmacological Insights into Autophagy Modulation in Cancer Treatment and Future Progress.Cells. 2023 Jan 31;12(3):458. doi: 10.3390/cells12030458. Cells. 2023. PMID: 36766800 Free PMC article. Review.
-
The Essential Oil from Conyza bonariensis (L.) Cronquist (Asteraceae) Exerts an In Vitro Antimelanoma Effect by Inducing Apoptosis and Modulating the MAPKs, NF-κB, and PKB/AKT Signaling Pathways.Pharmaceuticals (Basel). 2023 Nov 2;16(11):1553. doi: 10.3390/ph16111553. Pharmaceuticals (Basel). 2023. PMID: 38004419 Free PMC article.
-
Overcoming the therapeutic resistance of hepatomas by targeting the tumor microenvironment.Front Oncol. 2022 Nov 15;12:988956. doi: 10.3389/fonc.2022.988956. eCollection 2022. Front Oncol. 2022. PMID: 36457492 Free PMC article. Review.
-
Using Human 'Personalized' Cybrids to Identify Drugs/Agents That Can Regulate Chronic Lymphoblastic Leukemia Mitochondrial Dysfunction.Int J Mol Sci. 2023 Jul 3;24(13):11025. doi: 10.3390/ijms241311025. Int J Mol Sci. 2023. PMID: 37446202 Free PMC article.
References
-
- Assaraf Y.G., Brozovic A., Goncalves A.C., Jurkovicova D., Line A., Machuqueiro M., Saponara S., Sarmento-Ribeiro A.B., Xavier C.P.R., Vasconcelos M.H. The multi-factorial nature of clinical multidrug resistance in cancer. Drug Resist. Updates. 2019;46:100645. doi: 10.1016/j.drup.2019.100645. - DOI - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
