

CDK6 Degradation Is Counteracted by p16INK4A and p18INK4C in AML
- PMID: 35326705
- PMCID: PMC8946512
- DOI: 10.3390/cancers14061554
CDK6 Degradation Is Counteracted by p16INK4A and p18INK4C in AML
Abstract
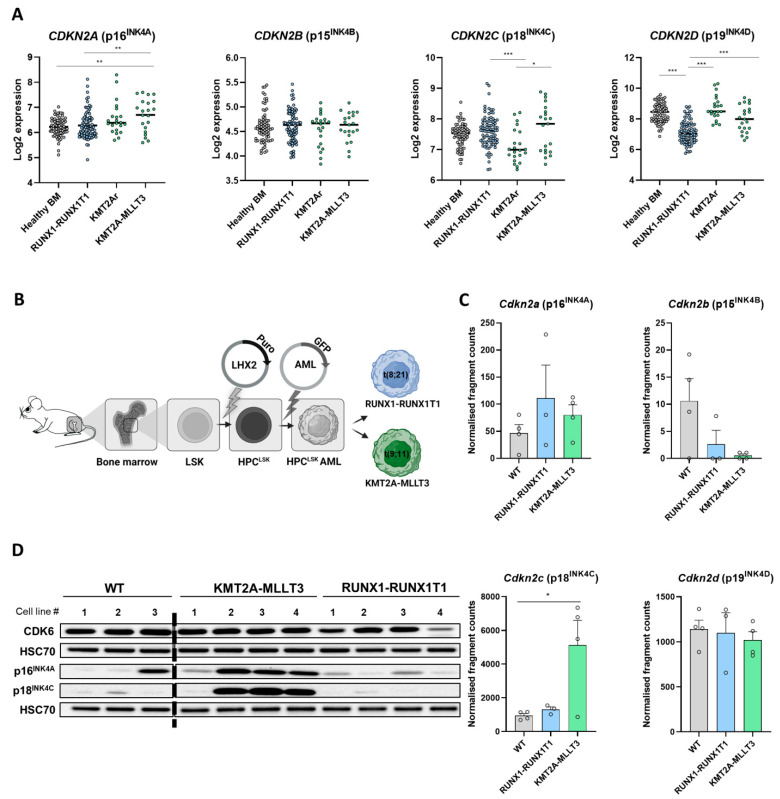
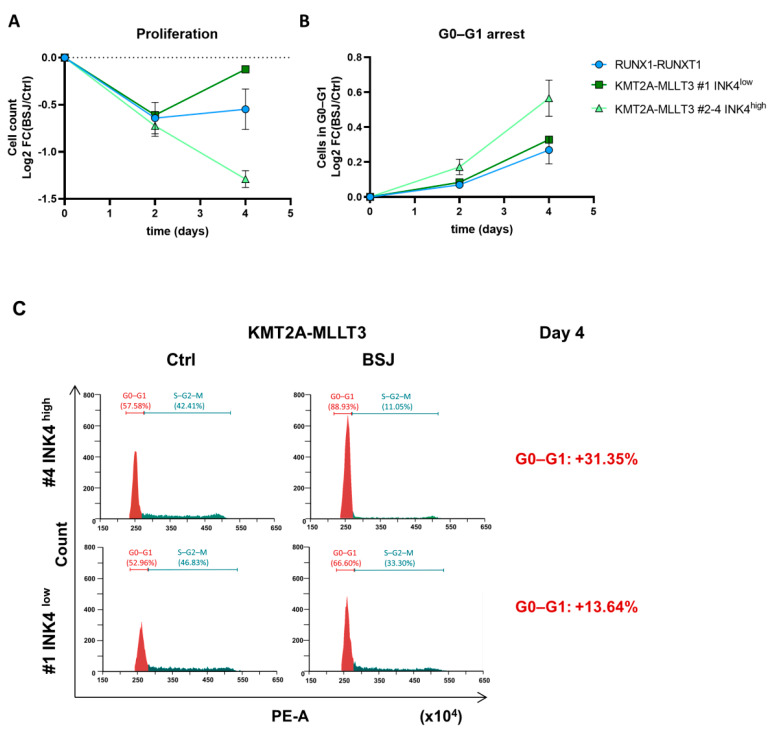
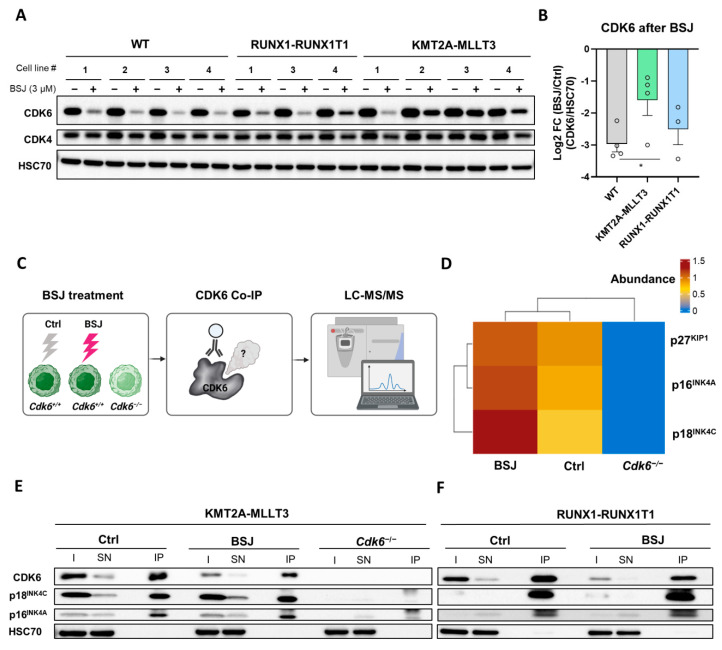
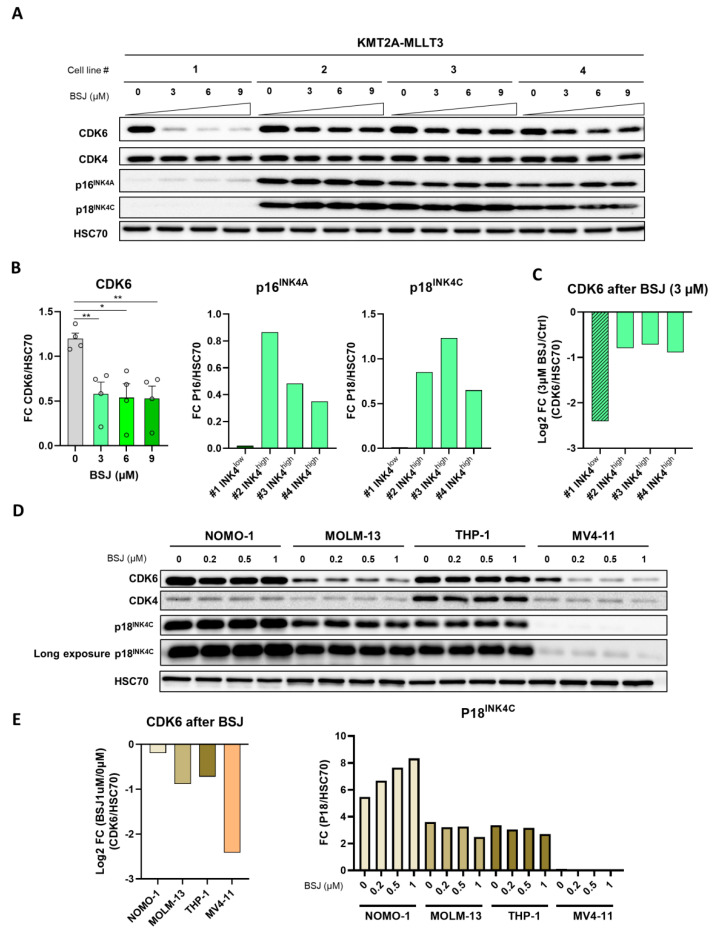
Cyclin-dependent kinase 6 (CDK6) represents a novel therapeutic target for the treatment of certain subtypes of acute myeloid leukaemia (AML). CDK4/6 kinase inhibitors have been widely studied in many cancer types and their effects may be limited by primary and secondary resistance mechanisms. CDK4/6 degraders, which eliminate kinase-dependent and kinase-independent effects, have been suggested as an alternative therapeutic option. We show that the efficacy of the CDK6-specific protein degrader BSJ-03-123 varies among AML subtypes and depends on the low expression of the INK4 proteins p16INK4A and p18INK4C. INK4 protein levels are significantly elevated in KMT2A-MLLT3+ cells compared to RUNX1-RUNX1T1+ cells, contributing to the different CDK6 degradation efficacy. We demonstrate that CDK6 complexes containing p16INK4A or p18INK4C are protected from BSJ-mediated degradation and that INK4 levels define the proliferative response to CDK6 degradation. These findings define INK4 proteins as predictive markers for CDK6 degradation-targeted therapies in AML.
Keywords: AML; CDK6; Cdkn2a; Cdkn2c; INK4; degrader; p16; p18.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Meyers R.M., Bryan J.G., McFarland J.M., Weir B.A., Sizemore A.E., Xu H., Dharia N.V., Montgomery P.G., Cowley G.S., Pantel S., et al. Computational correction of copy number effect improves specificity of CRISPR-Cas9 essentiality screens in cancer cells. Nat. Genet. 2017;49:1779–1784. doi: 10.1038/ng.3984. - DOI - PMC - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
