

Reactive Oxygen Species Induced Pathways in Heart Failure Pathogenesis and Potential Therapeutic Strategies
- PMID: 35327404
- PMCID: PMC8945343
- DOI: 10.3390/biomedicines10030602
Reactive Oxygen Species Induced Pathways in Heart Failure Pathogenesis and Potential Therapeutic Strategies
Abstract
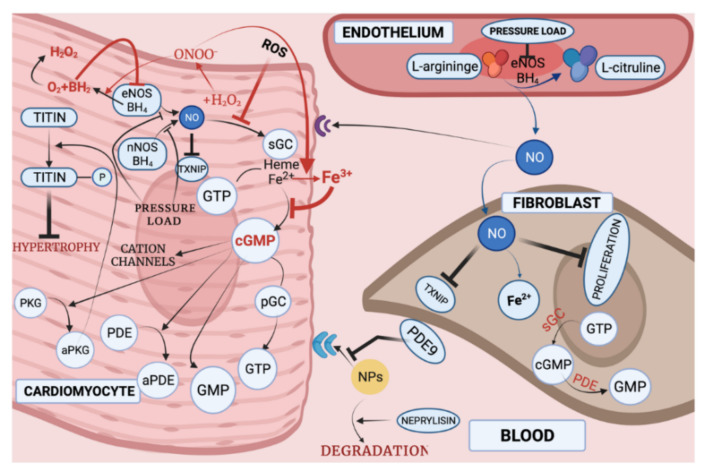
With respect to structural and functional cardiac disorders, heart failure (HF) is divided into HF with reduced ejection fraction (HFrEF) and HF with preserved ejection fraction (HFpEF). Oxidative stress contributes to the development of both HFrEF and HFpEF. Identification of a broad spectrum of reactive oxygen species (ROS)-induced pathways in preclinical models has provided new insights about the importance of ROS in HFrEF and HFpEF development. While current treatment strategies mostly concern neuroendocrine inhibition, recent data on ROS-induced metabolic pathways in cardiomyocytes may offer additional treatment strategies and targets for both of the HF forms. The purpose of this article is to summarize the results achieved in the fields of: (1) ROS importance in HFrEF and HFpEF pathophysiology, and (2) treatments for inhibiting ROS-induced pathways in HFrEF and HFpEF patients. ROS-producing pathways in cardiomyocytes, ROS-activated pathways in different HF forms, and treatment options to inhibit their action are also discussed.
Keywords: NO; cGC; heart failure with preserved ejection fraction; heart failure with reduced ejection fraction; protein kinases; reactive oxygen species.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
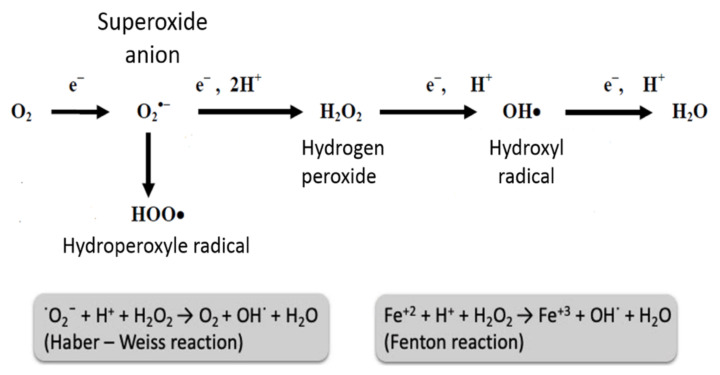
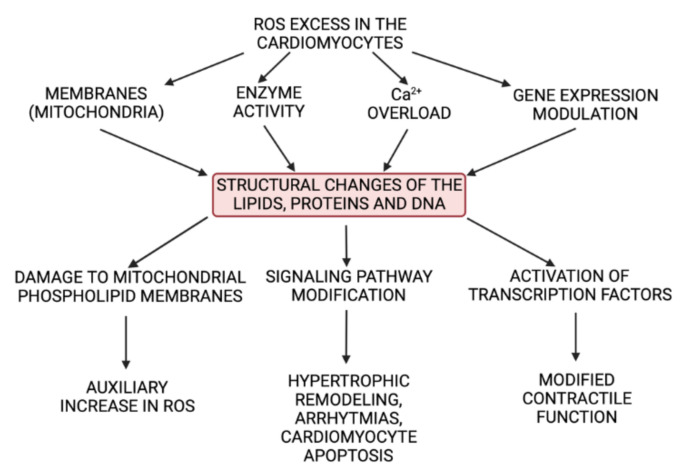
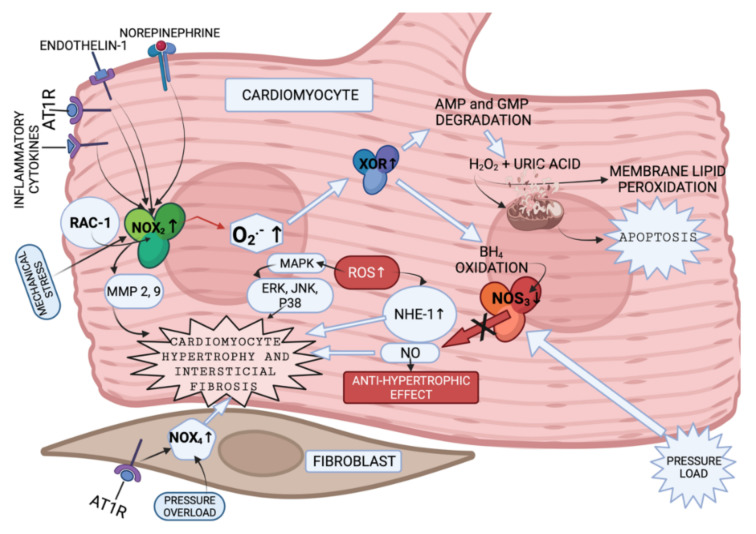
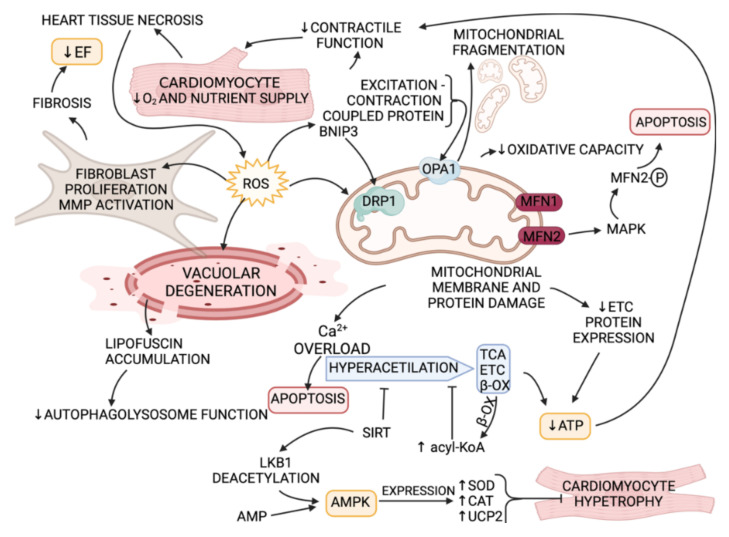
References
-
- Ponikowski P., Voors A.A., Anker S.D., Bueno H., Cleland J.G.F., Coats A.J.S. 2016 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure: The Task Force for the diagnosis and treatment of acute and chronic heart failure of the European Society of Cardiology (ESC)Developed with the special contribution of. Eur. Heart J. 2016;37:2129–2200. doi: 10.1093/eurheartj/ehw128. - DOI - PubMed
Publication types
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
