

Cutting-Edge Therapies and Novel Strategies for Acute Intermittent Porphyria: Step-by-Step towards the Solution
- PMID: 35327450
- PMCID: PMC8945550
- DOI: 10.3390/biomedicines10030648
Cutting-Edge Therapies and Novel Strategies for Acute Intermittent Porphyria: Step-by-Step towards the Solution
Abstract
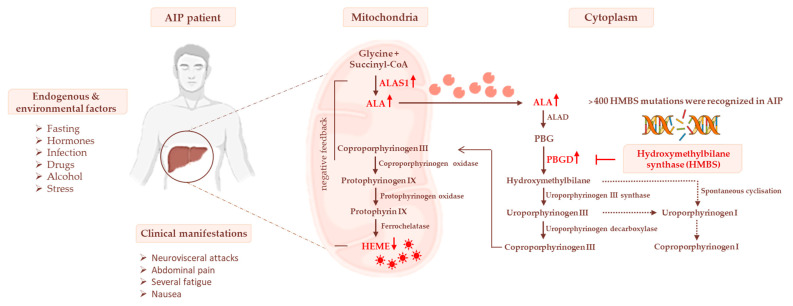
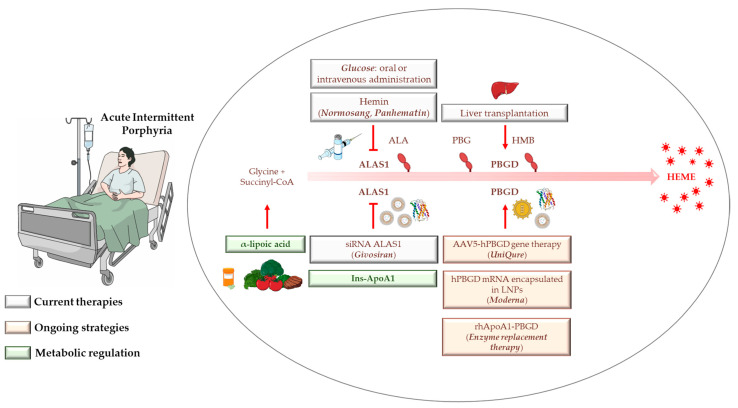
Acute intermittent porphyria (AIP) is an autosomal dominant disease caused by the hepatic deficiency of porphobilinogen deaminase (PBGD) and the slowdown of heme biosynthesis. AIP symptomatology includes life-threatening, acute neurovisceral or neuropsychiatric attacks manifesting in response to precipitating factors. The latter promote the upregulation of 5-aminolevulinic acid synthase-1 (ALAS1), the first enzyme of heme biosynthesis, which promotes the overload of neurotoxic porphyrin precursors. Hemin or glucose infusions are the first-line therapies for the reduction of ALAS1 levels in patients with mild to severe AIP, while liver transplantation is the only curative treatment for refractory patients. Recently, the RNA-interference against ALAS1 was approved as a treatment for adult and adolescent patients with AIP. These emerging therapies aim to substitute dysfunctional PBGD with adeno-associated vectors for genome editing, human PBGD mRNA encapsulated in lipid nanoparticles, or PBGD protein linked to apolipoprotein A1. Finally, the impairment of glucose metabolism linked to insulin resistance, and mitochondrial aberrations during AIP pathophysiology provided new therapeutic targets. Therefore, the use of liver-targeted insulin and insulin-mimetics such as α-lipoic acid may be useful for overcoming metabolic dysfunction in these subjects. Herein, the present review aims to provide an overview of AIP pathophysiology and management, focusing on conventional and recent therapeutical approaches.
Keywords: AIP; PBGD; heme; insulin; liver metabolism; α-lipoic acid.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
Similar articles
-
Phase I open label liver-directed gene therapy clinical trial for acute intermittent porphyria.J Hepatol. 2016 Oct;65(4):776-783. doi: 10.1016/j.jhep.2016.05.012. Epub 2016 May 17. J Hepatol. 2016. PMID: 27212246 Clinical Trial.
-
Understanding Carbohydrate Metabolism and Insulin Resistance in Acute Intermittent Porphyria.Int J Mol Sci. 2022 Dec 20;24(1):51. doi: 10.3390/ijms24010051. Int J Mol Sci. 2022. PMID: 36613492 Free PMC article. Review.
-
Systemic messenger RNA replacement therapy is effective in a novel clinically relevant model of acute intermittent porphyria developed in non-human primates.Gut. 2025 Jan 17;74(2):270-283. doi: 10.1136/gutjnl-2024-332619. Gut. 2025. PMID: 39366725 Free PMC article.
-
Therapeutic strategies for acute intermittent porphyria.Intractable Rare Dis Res. 2020 Nov;9(4):205-216. doi: 10.5582/irdr.2020.03089. Intractable Rare Dis Res. 2020. PMID: 33139979 Free PMC article. Review.
-
Porphobilinogen deaminase over-expression in hepatocytes, but not in erythrocytes, prevents accumulation of toxic porphyrin precursors in a mouse model of acute intermittent porphyria.J Hepatol. 2010 Mar;52(3):417-24. doi: 10.1016/j.jhep.2009.09.003. Epub 2009 Sep 23. J Hepatol. 2010. PMID: 19815305
Cited by
-
A case report of acute intermittent porphyria leading to severe disability.Front Neurol. 2024 Jan 11;14:1334743. doi: 10.3389/fneur.2023.1334743. eCollection 2023. Front Neurol. 2024. PMID: 38274883 Free PMC article.
-
The Alpha-Lipoic Acid Improves Glucose Metabolism and Hyperinsulinemia in Acute Intermittent Porphyria: A Nutritional Concept for the Management of Rare Disorders.Cell Mol Gastroenterol Hepatol. 2024;17(3):511-514. doi: 10.1016/j.jcmgh.2023.11.007. Epub 2023 Nov 17. Cell Mol Gastroenterol Hepatol. 2024. PMID: 37979725 Free PMC article. No abstract available.
-
Nanomaterials Based on 2,7,12,17-Tetra-tert-butyl-5,10,15,20-tetraaza-21H,23H-porphine Exhibiting Bifunctional Sensitivity for Monitoring Chloramphenicol and Co2.Biomedicines. 2024 Mar 30;12(4):770. doi: 10.3390/biomedicines12040770. Biomedicines. 2024. PMID: 38672126 Free PMC article.
-
[Abdominal pain and severely impaired consciousness in a 19-year-old female patient].Inn Med (Heidelb). 2025 Jul 16. doi: 10.1007/s00108-025-01939-9. Online ahead of print. Inn Med (Heidelb). 2025. PMID: 40668375 Review. German.
-
Understanding Hepatic Porphyrias: Symptoms, Treatments, and Unmet Needs.Semin Liver Dis. 2024 May;44(2):209-225. doi: 10.1055/s-0044-1787076. Epub 2024 May 17. Semin Liver Dis. 2024. PMID: 38772406 Free PMC article. Review.
References
-
- Bonkovsky H.L., Maddukuri V.C., Yazici C., Anderson K.E., Bissell D.M., Bloomer J.R., Phillips J.D., Naik H., Peter I., Baillargeon G., et al. Acute porphyrias in the USA: Features of 108 subjects from porphyrias consortium. Am. J. Med. 2014;127:1233–1241. doi: 10.1016/j.amjmed.2014.06.036. - DOI - PMC - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
