

In Silico Exploration of Mycobacterium tuberculosis Metabolic Networks Shows Host-Associated Convergent Fluxomic Phenotypes
- PMID: 35327567
- PMCID: PMC8945471
- DOI: 10.3390/biom12030376
In Silico Exploration of Mycobacterium tuberculosis Metabolic Networks Shows Host-Associated Convergent Fluxomic Phenotypes
Abstract
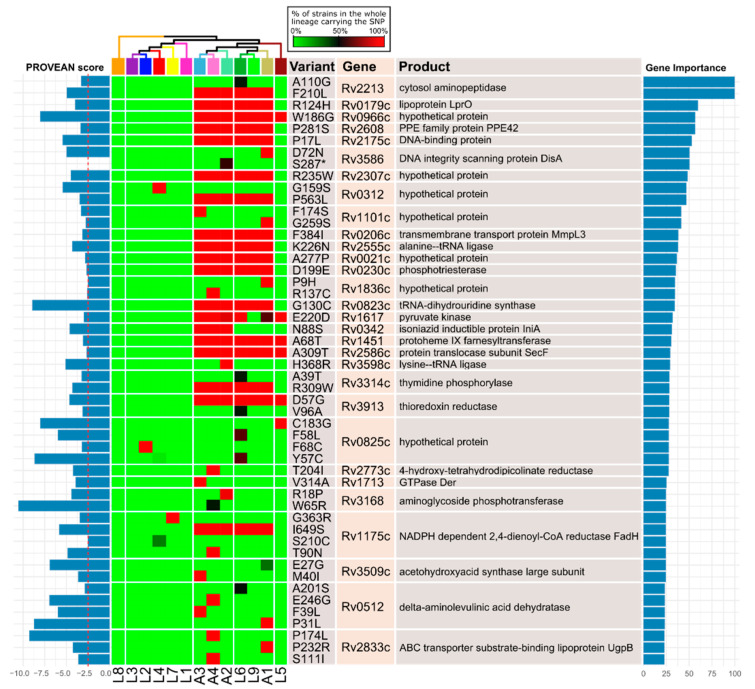
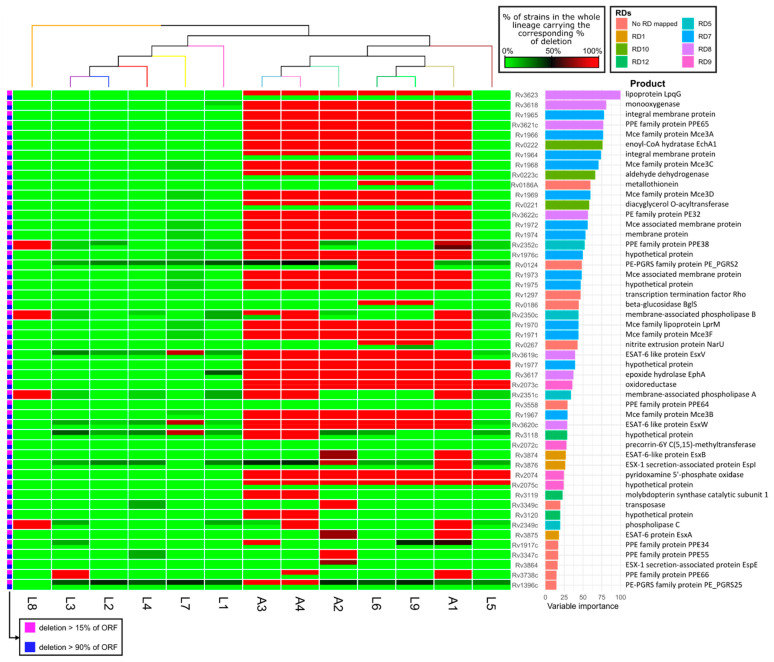
Mycobacterium tuberculosis, the causative agent of tuberculosis, is composed of several lineages characterized by a genome identity higher than 99%. Although the majority of the lineages are associated with humans, at least four lineages are adapted to other mammals, including different M. tuberculosis ecotypes. Host specificity is associated with higher virulence in its preferred host in ecotypes such as M. bovis. Deciphering what determines the preference of the host can reveal host-specific virulence patterns. However, it is not clear which genomic determinants might be influencing host specificity. In this study, we apply a combination of unsupervised and supervised classification methods on genomic data of ~27,000 M. tuberculosis clinical isolates to decipher host-specific genomic determinants. Host-specific genomic signatures are scarce beyond known lineage-specific mutations. Therefore, we integrated lineage-specific mutations into the iEK1011 2.0 genome-scale metabolic model to obtain lineage-specific versions of it. Flux distributions sampled from the solution spaces of these models can be accurately separated according to host association. This separation correlated with differences in cell wall processes, lipid, amino acid and carbon metabolic subsystems. These differences were observable when more than 95% of the samples had a specific growth rate significantly lower than the maximum achievable by the models. This suggests that these differences might manifest at low growth rate settings, such as the restrictive conditions M. tuberculosis suffers during macrophage infection.
Keywords: Mycobacterium tuberculosis; genome-scale metabolic model; host association; lineage; metabolic networks.
Conflict of interest statement
The authors declare no conflict of interest.
Figures





References
-
- WHO . Global Tuberculosis Report 2021. WHO; Geneva, Switzerland: 2021.
-
- Brites D., Gagneux S. Strain Variation in the Mycobacterium tuberculosis Complex: Its Role in Biology, Epidemiology and Control. Springer; Cham, Switzerland: 2017. The Nature and Evolution of Genomic Diversity in the Mycobacterium Tuberculosis Complex; pp. 1–26. - PubMed
-
- Yeboah-Manu D., de Jong B.C., Gehre F. Strain Variation in the Mycobacterium tuberculosis Complex: Its Role in Biology, Epidemiology and Control. Springer; Cham, Switzerland: 2017. The Biology and Epidemiology of Mycobacterium Africanum; pp. 117–133. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Medical
