

Genetic and Clinical Studies of Peripheral Neuropathies with Three Small Heat Shock Protein Gene Variants in Korea
- PMID: 35328016
- PMCID: PMC8949397
- DOI: 10.3390/genes13030462
Genetic and Clinical Studies of Peripheral Neuropathies with Three Small Heat Shock Protein Gene Variants in Korea
Abstract
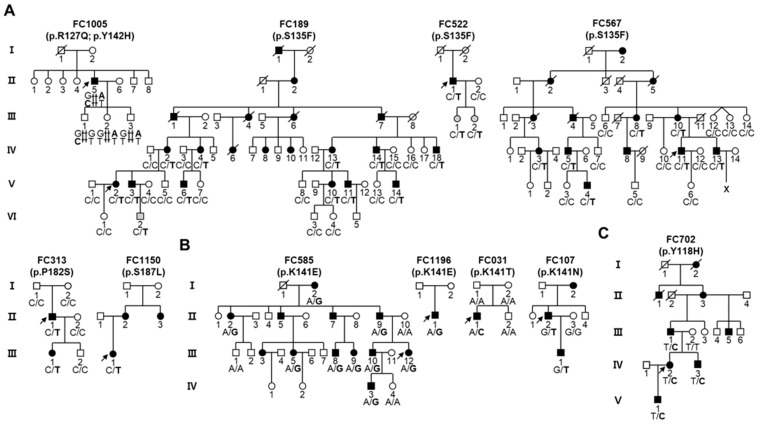
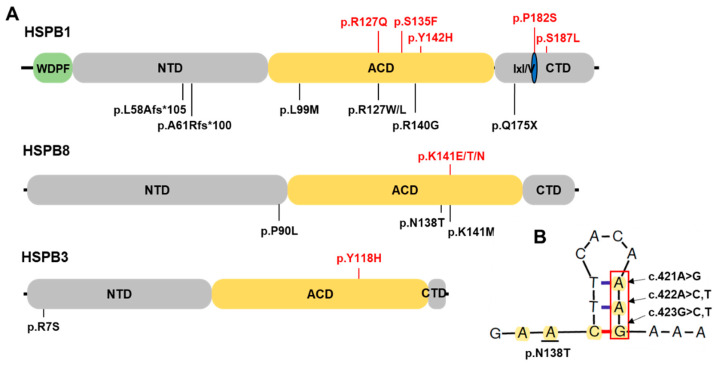
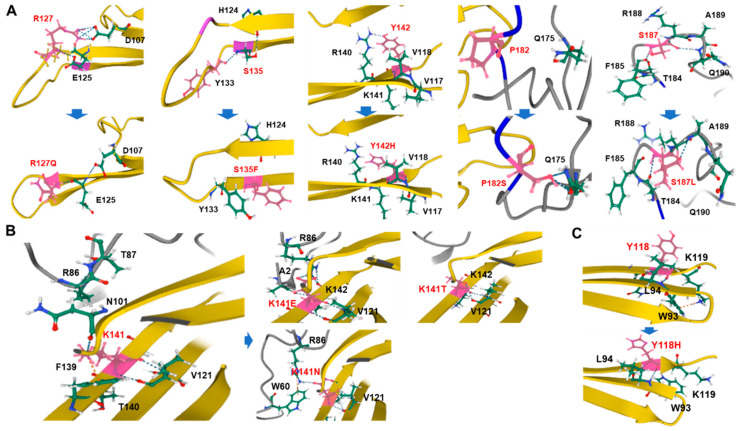
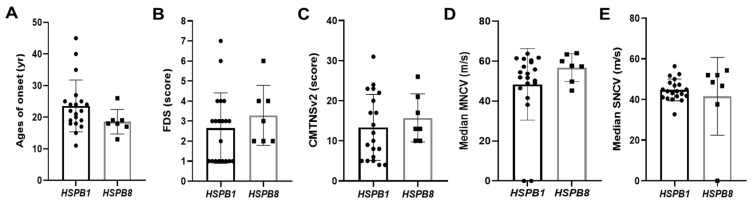
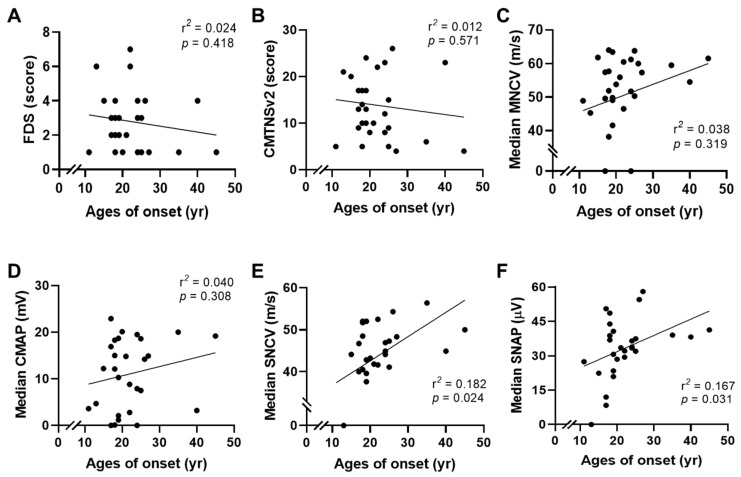
Small heat shock proteins (sHSPs) are ATP-independent chaperones that help correct the folding of denatured proteins and protect cells from stress. Mutations in HSPB1, HSPB8, and HSPB3 are implicated in inherited peripheral neuropathies (IPNs), such as Charcot-Marie-Tooth disease type 2 (CMT2) and distal hereditary motor neuropathies (dHMN). This study, using whole exome sequencing or targeted gene sequencing, identified 9 pathogenic or likely pathogenic variants in these three sHSP genes from 11 Korean IPN families. Most variants were located in the evolutionally well conserved α-crystallin domain, except for p.P182S and p.S187L in HSPB1. As an atypical case, a patient with dHMN2 showed two compound heterozygous variants of p.R127Q and p.Y142H in HSPB1, suggesting a putative case of recessive inheritance, which requires additional research to confirm. Three HSPB8 variants were located in the p.K141 residue, which seemed to be a mutational hot spot. There were no significant differences between patient groups, which divided by sHSP genes for clinical symptoms such as onset age, severity, and nerve conduction. Early-onset patients showed a tendency of slightly decreased sensory nerve conduction values compared with late-onset patients. As a first Korean IPN cohort study examining sHSP genes, these results will, we believe, be helpful for molecular diagnosis and care of patients with CMT2 and dHMN.
Keywords: Charcot-Marie-Tooth disease type 2; HSPB1; HSPB3; HSPB8; Korean; distal hereditary motor neuropathies.
Conflict of interest statement
The authors declare no conflict of interest.
Figures

References
-
- Evgrafov O.V., Mersiyanova I., Irobi J., Van Den Bosch L., Dierick I., Leung C.L., Schagina O., Verpoorten N., Van Impe K., Fedotov V., et al. Mutant small heat-shock protein 27 causes axonal Charcot-Marie-Tooth disease and distal hereditary motor neuropathy. Nat. Genet. 2004;36:602–606. doi: 10.1038/ng1354. - DOI - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
