

Cell Signaling Pathways That Promote Radioresistance of Cancer Cells
- PMID: 35328212
- PMCID: PMC8947583
- DOI: 10.3390/diagnostics12030656
Cell Signaling Pathways That Promote Radioresistance of Cancer Cells
Abstract
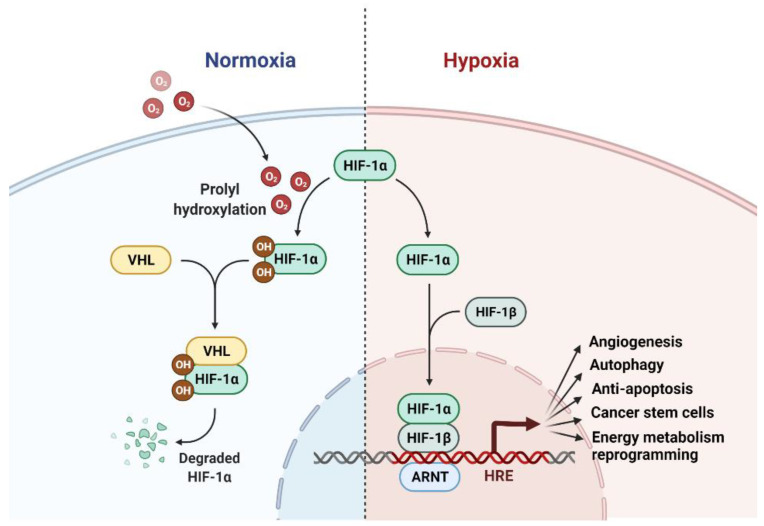
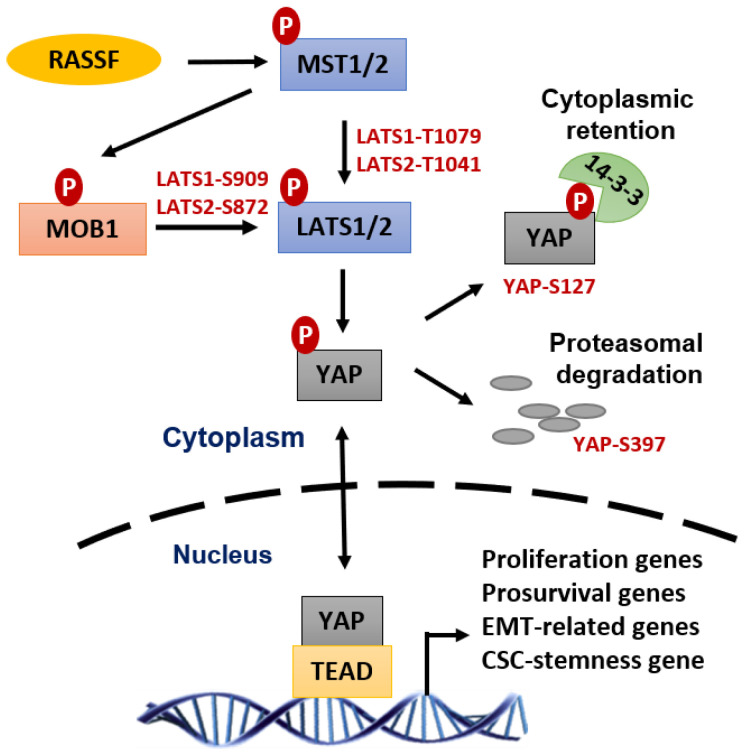
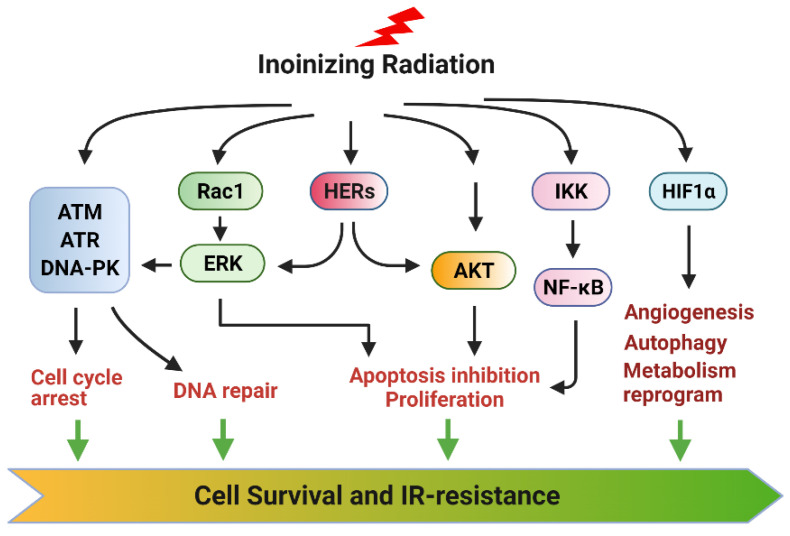
Radiation therapy (RT) is a standard treatment for solid tumors and about 50% of patients with cancer, including pediatric cancer, receive RT. While RT has significantly improved the overall survival and quality of life of cancer patients, its efficacy has still been markedly limited by radioresistance in a significant number of cancer patients (intrinsic or acquired), resulting in failure of the RT control of the disease. Radiation eradicates cancer cells mainly by causing DNA damage. However, radiation also concomitantly activates multiple prosurvival signaling pathways, which include those mediated by ATM, ATR, AKT, ERK, and NF-κB that promote DNA damage checkpoint activation/DNA repair, autophagy induction, and/or inhibition of apoptosis. Furthermore, emerging data support the role of YAP signaling in promoting the intrinsic radioresistance of cancer cells, which occurs through its activation of the transcription of many essential genes that support cell survival, DNA repair, proliferation, and the stemness of cancer stem cells. Together, these signaling pathways protect cancer cells by reducing the magnitude of radiation-induced cytotoxicity and promoting radioresistance. Thus, targeting these prosurvival signaling pathways could potentially improve the radiosensitivity of cancer cells. In this review, we summarize the contribution of these pathways to the radioresistance of cancer cells.
Keywords: DNA repair; apoptosis; autophagy; cell cycle checkpoint; cell signaling pathways; radiation therapy.
Conflict of interest statement
The authors declare that they have no competing interest.
Figures
Similar articles
-
Radiation-induced signaling pathways that promote cancer cell survival (review).Int J Oncol. 2014 Nov;45(5):1813-9. doi: 10.3892/ijo.2014.2614. Epub 2014 Aug 20. Int J Oncol. 2014. PMID: 25174607 Free PMC article. Review.
-
Inhibition of radiation-induced DNA repair and prosurvival pathways contributes to vorinostat-mediated radiosensitization of pancreatic cancer cells.Pancreas. 2010 Nov;39(8):1277-83. doi: 10.1097/MPA.0b013e3181dd63e1. Pancreas. 2010. PMID: 20531243 Free PMC article.
-
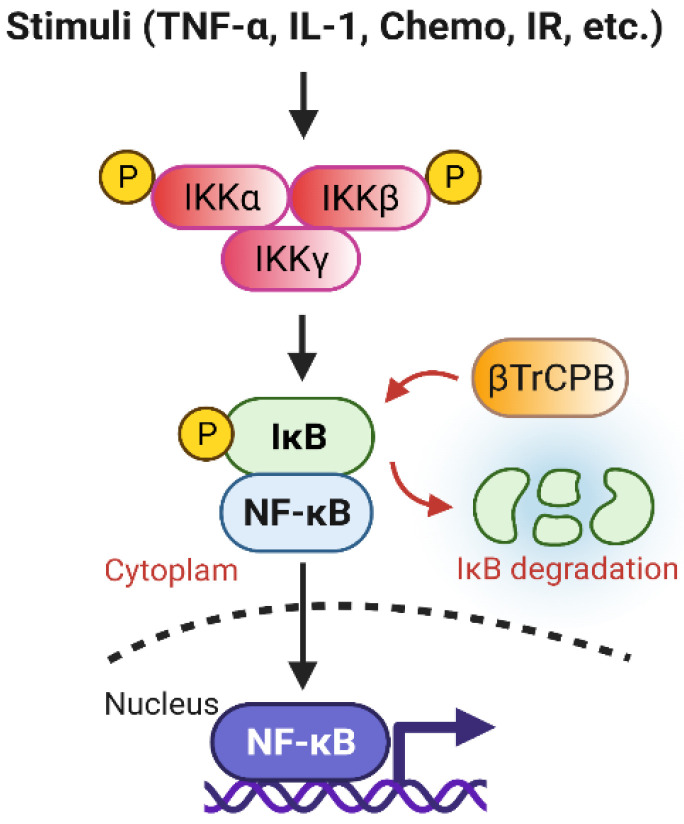
The Accomplices of NF-κB Lead to Radioresistance.Curr Protein Pept Sci. 2015;16(4):279-94. doi: 10.2174/138920371604150429152328. Curr Protein Pept Sci. 2015. PMID: 25929862 Review.
-
Activation of the phosphorylation of ATM contributes to radioresistance of glioma stem cells.Oncol Rep. 2013 Oct;30(4):1793-801. doi: 10.3892/or.2013.2614. Epub 2013 Jul 11. Oncol Rep. 2013. PMID: 23846672
-
Targeting the AKT/GSK3β/cyclin D1/Cdk4 survival signaling pathway for eradication of tumor radioresistance acquired by fractionated radiotherapy.Int J Radiat Oncol Biol Phys. 2011 Jun 1;80(2):540-8. doi: 10.1016/j.ijrobp.2010.12.065. Epub 2011 Mar 11. Int J Radiat Oncol Biol Phys. 2011. PMID: 21398050
Cited by
-
Non-coding RNAs as modulators of radioresponse in triple-negative breast cancer: a systematic review.J Biomed Sci. 2024 Oct 2;31(1):93. doi: 10.1186/s12929-024-01081-y. J Biomed Sci. 2024. PMID: 39354523 Free PMC article.
-
Understanding the molecular mechanism responsible for developing therapeutic radiation-induced radioresistance of rectal cancer and improving the clinical outcomes of radiotherapy - A review.Cancer Biol Ther. 2024 Dec 31;25(1):2317999. doi: 10.1080/15384047.2024.2317999. Epub 2024 Mar 6. Cancer Biol Ther. 2024. PMID: 38445632 Free PMC article. Review.
-
Bladder Cancer Treatments in the Age of Personalized Medicine: A Comprehensive Review of Potential Radiosensitivity Biomarkers.Biomark Insights. 2024 Nov 6;19:11772719241297168. doi: 10.1177/11772719241297168. eCollection 2024. Biomark Insights. 2024. PMID: 39512649 Free PMC article. Review.
-
Emerging Role of Epigenetic Modifiers in Breast Cancer Pathogenesis and Therapeutic Response.Cancers (Basel). 2023 Aug 7;15(15):4005. doi: 10.3390/cancers15154005. Cancers (Basel). 2023. PMID: 37568822 Free PMC article. Review.
-
RNA Binding Proteins are Pivotal Regulators of Cancer Radioresistance and Potential Targets for Preventing Tumor Recurrence.Curr Radiopharm. 2025;18(4):245-261. doi: 10.2174/0118744710366175250425101010. Curr Radiopharm. 2025. PMID: 40304321 Review.
References
-
- Wilkinson-Ryan I., Binder P.S., Pourabolghasem S., Al-Hammadi N., Fuh K., Hagemann A., Thaker P., Schwarz J., Grigsby P., Mutch D., et al. Concomitant chemotherapy and radiation for the treatment of advanced-stage endometrial cancer. Gynecol. Oncol. 2014;134:24–28. doi: 10.1016/j.ygyno.2014.05.002. - DOI - PubMed
-
- White R.R., Xie H.B., Gottfried M.R., Czito B.G., Hurwitz H.I., Morse M.A., Blobe G.C., Paulson E.K., Baillie J., Branch M.S., et al. Significance of histological response to preoperative chemoradiotherapy for pancreatic cancer. Ann. Surg. Oncol. 2005;12:214–221. doi: 10.1245/ASO.2005.03.105. - DOI - PubMed
Publication types
Grants and funding
- Great Plains IDeA-CTR-Pilot Projects Program (5U54GM115458)/Pilot project award (no number)/GM/NIGMS NIH HHS/United States
- R01GM143329/GM/NIGMS NIH HHS/United States
- Nebraska/DHHS (2022-59)/Nebraska, Department of Health and Human Services
- W81XWH2110700/Congressionally Directed Medical Research Programs
- Nebraska/DHHS (2022-42)/Nebraska, Department of Health and Human Services
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
