

Helicobacter pylori Urease: Potential Contributions to Alzheimer's Disease
- PMID: 35328512
- PMCID: PMC8949269
- DOI: 10.3390/ijms23063091
Helicobacter pylori Urease: Potential Contributions to Alzheimer's Disease
Abstract
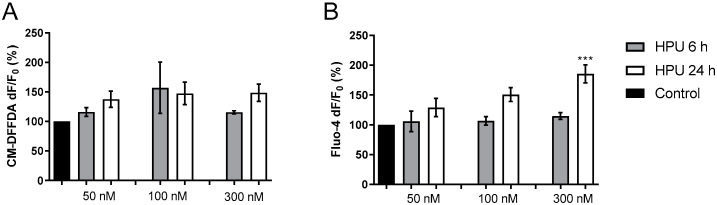
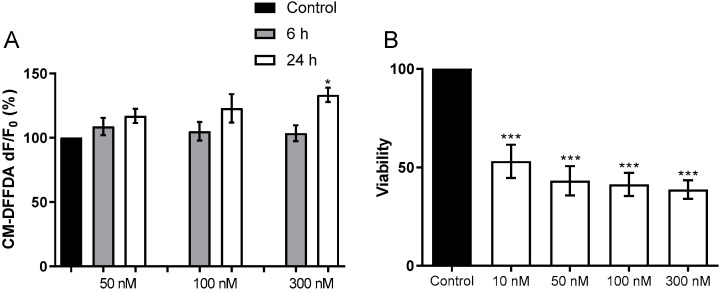
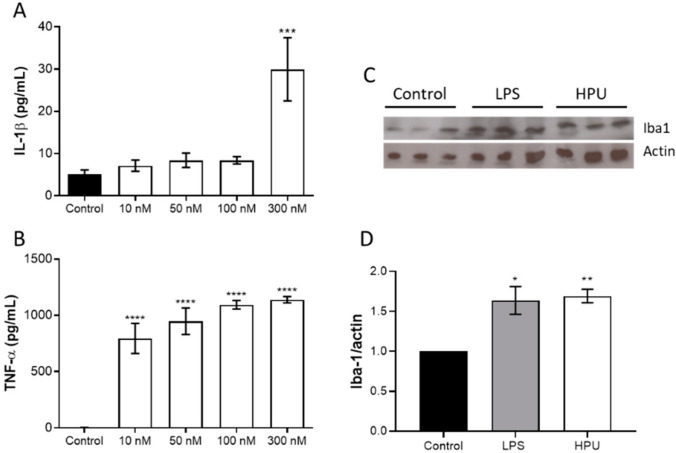
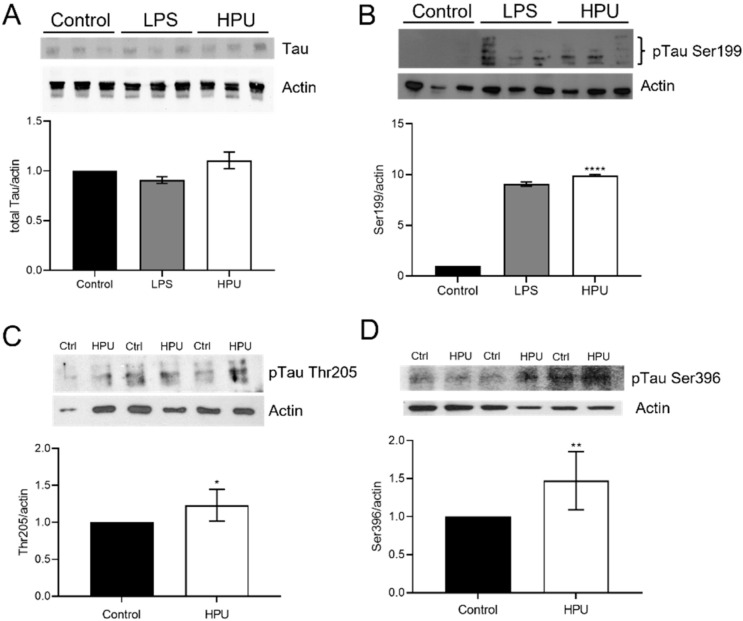
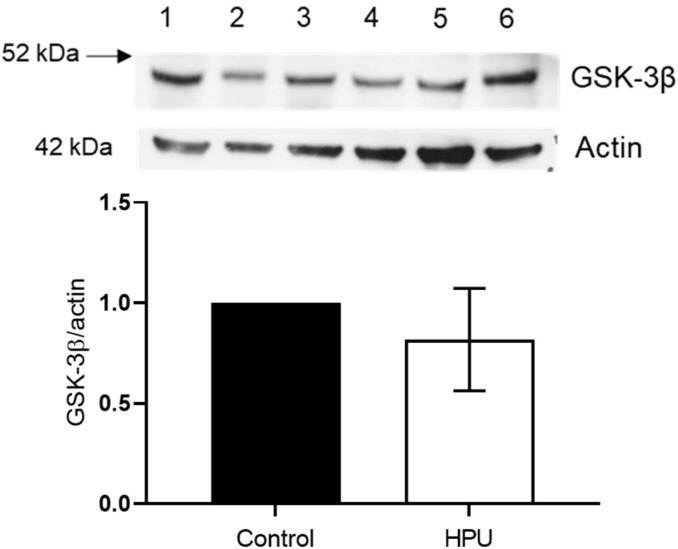
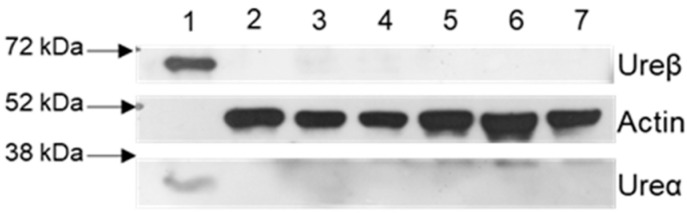
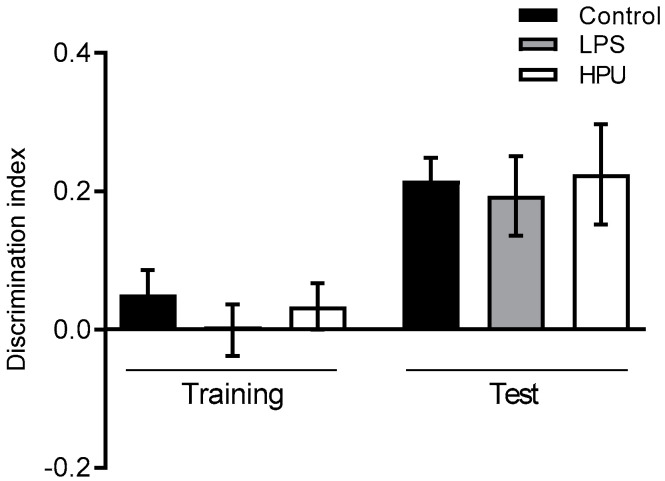
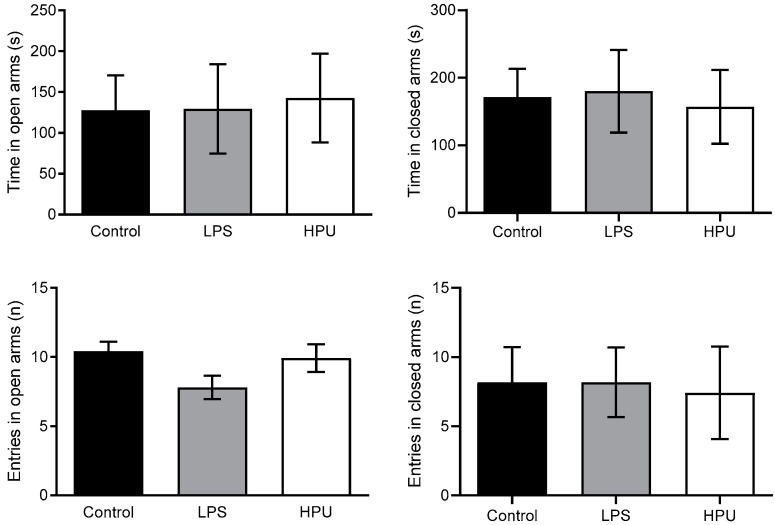
Alzheimer's disease (AD) causes dementia and memory loss in the elderly. Deposits of beta-amyloid peptide and hyperphosphorylated tau protein are present in a brain with AD. A filtrate of Helicobacter pylori's culture was previously found to induce hyperphosphorylation of tau in vivo, suggesting that bacterial exotoxins could permeate the blood-brain barrier and directly induce tau's phosphorylation. H. pylori, which infects ~60% of the world population and causes gastritis and gastric cancer, produces a pro-inflammatory urease (HPU). Here, the neurotoxic potential of HPU was investigated in cultured cells and in rats. SH-SY5Y neuroblastoma cells exposed to HPU (50-300 nM) produced reactive oxygen species (ROS) and had an increased [Ca2+]i. HPU-treated BV-2 microglial cells produced ROS, cytokines IL-1β and TNF-α, and showed reduced viability. Rats received daily i.p., HPU (5 µg) for 7 days. Hyperphosphorylation of tau at Ser199, Thr205 and Ser396 sites, with no alterations in total tau or GSK-3β levels, and overexpression of Iba1, a marker of microglial activation, were seen in hippocampal homogenates. HPU was not detected in the brain homogenates. Behavioral tests were performed to assess cognitive impairments. Our findings support previous data suggesting an association between infection by H. pylori and tauopathies such as AD, possibly mediated by its urease.
Keywords: BV-2 microglia; Helicobacter pylori; SH-SY5Y neuroblastoma cells; elevated plus maze; neuroinflammation; object recognition test; pro-inflammatory cytokines; tau hyperphosphorylation; urease.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Hooi J.K., Lai W.Y., Ng W.K., Suen M.M., Underwood F.E., Tanyingoh D., Malfertheiner P., Graham D.Y., Wong V.W., Wu J.C., et al. Global Prevalence of Helicobacter pylori Infection: Systematic Review and Meta-Analysis. Gastroenterology. 2017;153:420–429. doi: 10.1053/j.gastro.2017.04.022. - DOI - PubMed
MeSH terms
Substances
Grants and funding
- Finance code 001 and Edital Toxinologia - grant 63/2010/Coordenação de Aperfeicoamento de Pessoal de Nível Superior
- Edital Universal, grants 44.6052/2014-1 and 47.5908/2012-0/National Council for Scientific and Technological Development
- Ed. PPSUS, grant 17/2551-0001451-0/Fundação de Amparo à Pesquisa do Estado do Rio Grande do Sul
- National Institute of Science and Technology in Brain Diseases, Excitotoxicity and Neuroprotection - CNPq 46.5671/1-2014/National Council for Scientific and Technological Development
- National Institute of Science and Technology in Brain Diseases, Excitotoxicity and Neuroprotection - FAPERGS 17/2551-0000516-3/Fundação de Amparo à Pesquisa do Estado do Rio Grande do Sul
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
