

Genotoxic stress signalling as a driver of macrophage diversity
- PMID: 35330617
- PMCID: PMC8892193
- DOI: 10.15698/cst2022.03.265
Genotoxic stress signalling as a driver of macrophage diversity
Abstract
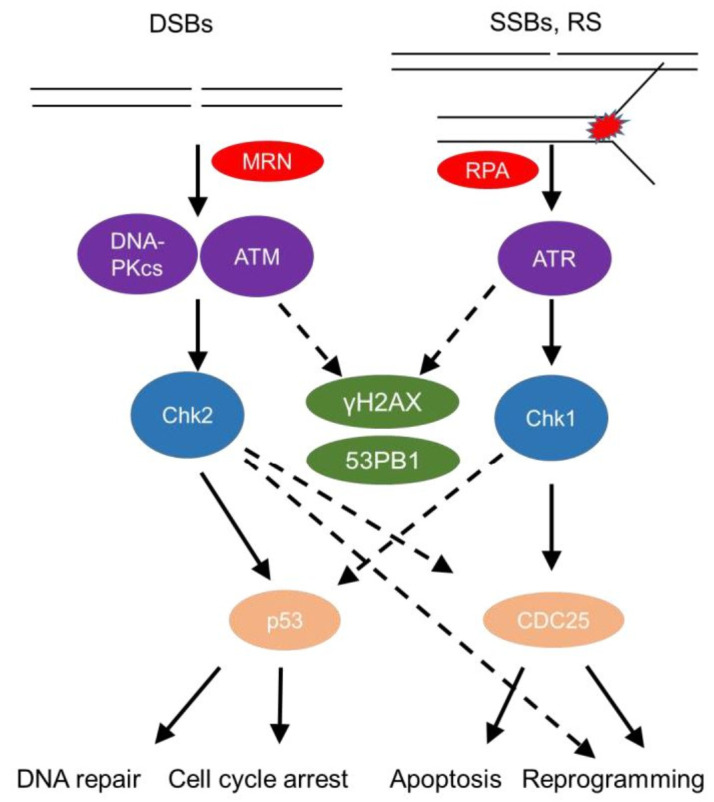
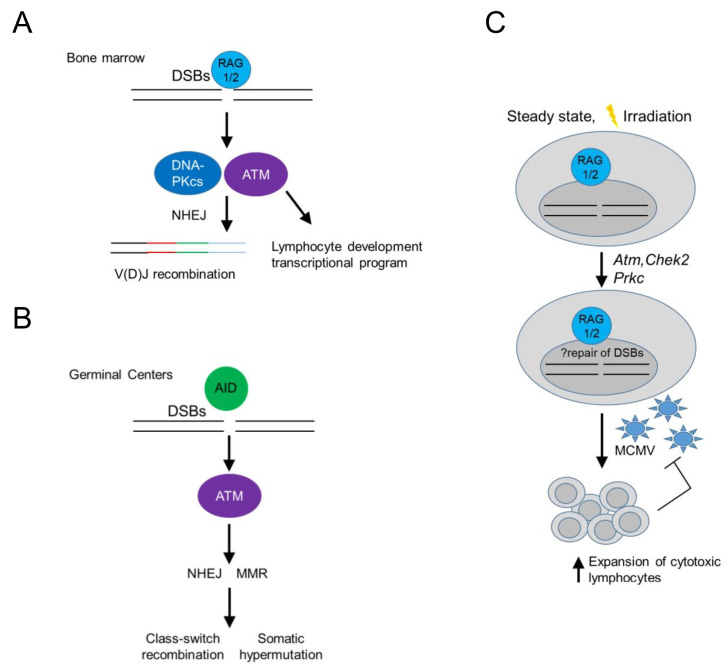
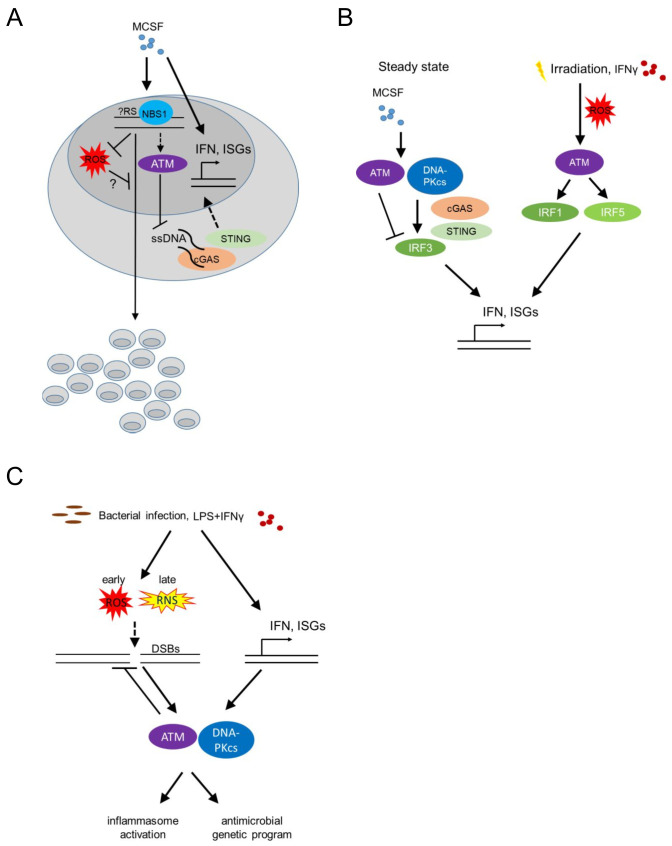
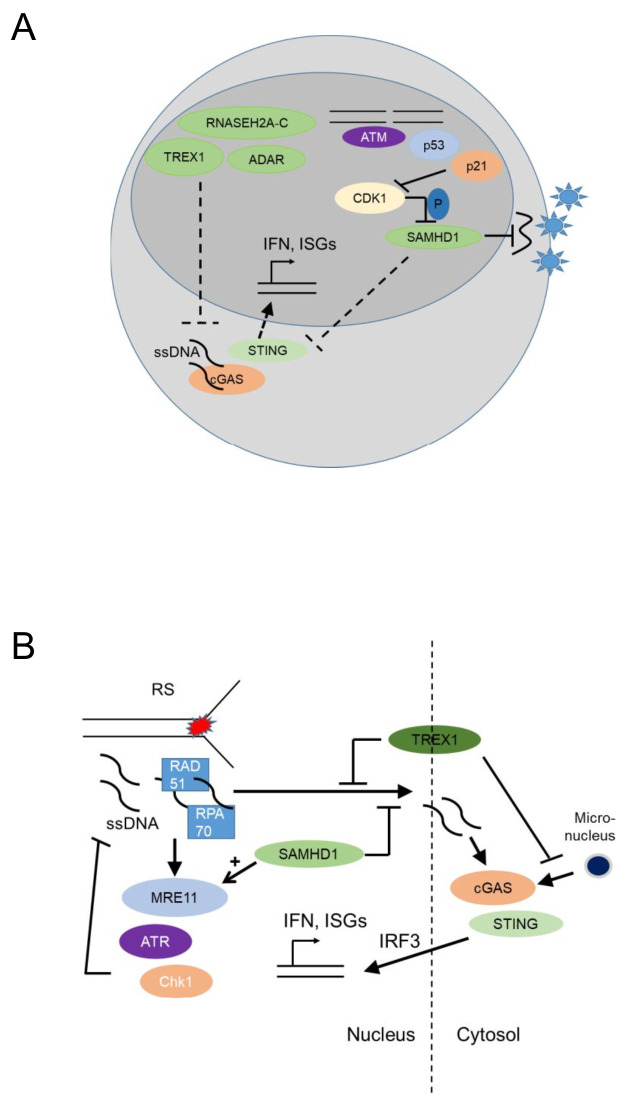
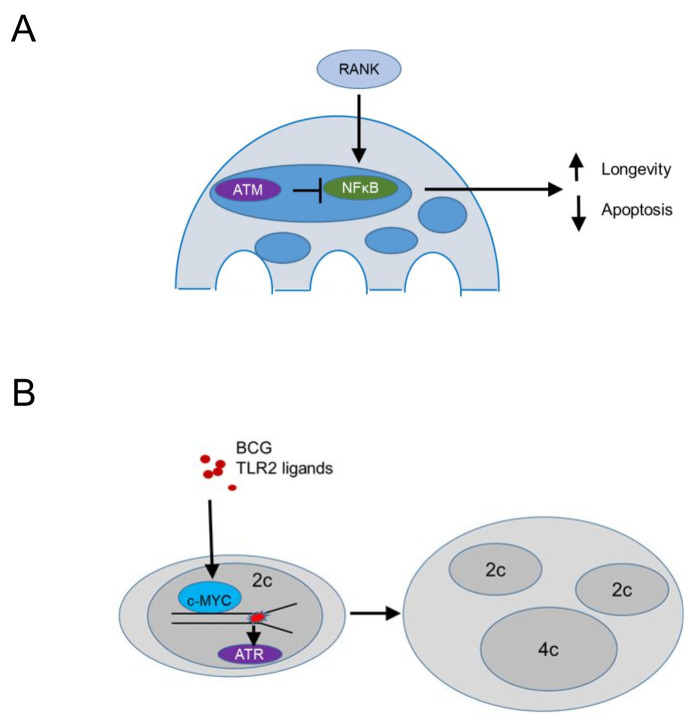
Tissue macrophages arise from yolk sac, fetal liver and hematopoietic progenitors and adopt diverse transcriptional programs and phenotypes, instructed by their microenvironment. In chronic inflammation, such as in chronic infections, autoimmunity, or cancer, tissue microenvironments change dramatically thus imprinting new programs on tissue macrophages. While stress is a known driver of carcinogenesis in epithelial cells, emerging evidence suggests that macrophage responses to genotoxic stress are embedded in their 'physiologic' immune and tissue healing programs and in most cases do not lead to myeloid malignancies. The role of genotoxic stress as an instructor of macrophage-mediated immune defense and tissue remodeling is only beginning to be understood. Here, we review the evidence showing that genotoxic stress, which macrophages and their precursors face upon encountering inflammatory and/or growth signals, instructs their transcriptional programs, by activating non-canonical, cell-type specific DNA Damage Response (DDR)-driven signaling pathways. We propose that immune-cell specific, DDR-instructed programs are crucial for tissue homeostasis as well as for the maintenance and resolution of inflammatory responses in infection, cancer, autoinflammatory and autoimmune microenvironments.
Keywords: ATM; ATR; DNA damage; chronic inflammation; innate immunity; macrophage programs.
Copyright: © 2022 Kasapi and Triantafyllopoulou.
Conflict of interest statement
Conflict of Interest: The authors certify that they have no affiliations with or involvement in any organization or entity with any financial or non-financial interest in the subject matter or materials discussed in this manuscript.
Figures
Similar articles
-
Fetal monocytes and the origins of tissue-resident macrophages.Cell Immunol. 2018 Aug;330:5-15. doi: 10.1016/j.cellimm.2018.01.001. Epub 2018 Jan 12. Cell Immunol. 2018. PMID: 29475558 Review.
-
Early hematopoiesis and macrophage development.Semin Immunol. 2015 Dec;27(6):379-87. doi: 10.1016/j.smim.2016.03.013. Epub 2016 Mar 25. Semin Immunol. 2015. PMID: 27021646 Free PMC article. Review.
-
DNA Damage Signaling Instructs Polyploid Macrophage Fate in Granulomas.Cell. 2016 Nov 17;167(5):1264-1280.e18. doi: 10.1016/j.cell.2016.09.054. Epub 2016 Oct 27. Cell. 2016. PMID: 28084216
-
Tissue microenvironments define and get reinforced by macrophage phenotypes in homeostasis or during inflammation, repair and fibrosis.J Innate Immun. 2012;4(5-6):463-77. doi: 10.1159/000336717. Epub 2012 Apr 11. J Innate Immun. 2012. PMID: 22507825 Free PMC article. Review.
-
DNA damage signaling and polyploid macrophages in chronic inflammation.Curr Opin Immunol. 2018 Feb;50:55-63. doi: 10.1016/j.coi.2017.11.002. Epub 2017 Dec 1. Curr Opin Immunol. 2018. PMID: 29202328 Review.
Cited by
-
[The role of the response to DNA damage in granulomatous diseases].Z Rheumatol. 2022 Dec;81(10):881-887. doi: 10.1007/s00393-022-01260-y. Epub 2022 Aug 25. Z Rheumatol. 2022. PMID: 36006470 Free PMC article. German.
-
Why Don't the Mutant Cells That Evade DNA Repair Cause Cancer More Frequently? Importance of the Innate Immune System in the Tumor Microenvironment.Int J Mol Sci. 2023 Mar 6;24(5):5026. doi: 10.3390/ijms24055026. Int J Mol Sci. 2023. PMID: 36902456 Free PMC article. Review.
-
Thioredoxin A of Streptococcus suis Serotype 2 Contributes to Virulence by Inhibiting the Expression of Pentraxin 3 to Promote Survival Within Macrophages.J Microbiol. 2023 Apr;61(4):433-448. doi: 10.1007/s12275-023-00038-4. Epub 2023 Apr 3. J Microbiol. 2023. PMID: 37010796
-
Unique structural configuration of EV-DNA primes Kupffer cell-mediated antitumor immunity to prevent metastatic progression.Nat Cancer. 2024 Dec;5(12):1815-1833. doi: 10.1038/s43018-024-00862-6. Epub 2024 Dec 3. Nat Cancer. 2024. PMID: 39627554 Free PMC article.
References
-
- Yona S, Kim KW, Wolf Y, Mildner A, Varol D, Breker M, Strauss-Ayali D, Viukov S, Guilliams M, Misharin A, Hume DA, Perlman H, Malissen B, Zelzer E, Jung S. Fate mapping reveals origins and dynamics of monocytes and tissue macrophages under homeostasis. Immunity. 2013;38(1):79–91. doi: 10.1016/j.immuni.2012.12.001. - DOI - PMC - PubMed
-
- Gomez Perdiguero E, Klapproth K, Schulz C, Busch K, Azzoni E, Crozet L, Garner H, Trouillet C, de Bruijn MF, Geissmann F, Rodewald HR. Tissue-resident macrophages originate from yolk-sac-derived erythro-myeloid progenitors. Nature. 2015;518(7540):547–551. doi: 10.1038/nature13989. - DOI - PMC - PubMed
-
- Hashimoto D, Chow A, Noizat C, Teo P, Beasley MB, Leboeuf M, Becker CD, See P, Price J, Lucas D, Greter M, Mortha A, Boyer SW, Forsberg EC, Tanaka M, van Rooijen N, García-Sastre A, Stanley ER, Ginhoux F, Frenette PS, Merad M. Tissue-resident macrophages self-maintain locally throughout adult life with minimal contribution from circulating monocytes. Immunity. 2013;38(4):792–804. doi: 10.1016/j.immuni.2013.04.004. - DOI - PMC - PubMed
Publication types
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous