

A hidden layer of structural variation in transposable elements reveals potential genetic modifiers in human disease-risk loci
- PMID: 35332097
- PMCID: PMC8997352
- DOI: 10.1101/gr.275515.121
A hidden layer of structural variation in transposable elements reveals potential genetic modifiers in human disease-risk loci
Abstract
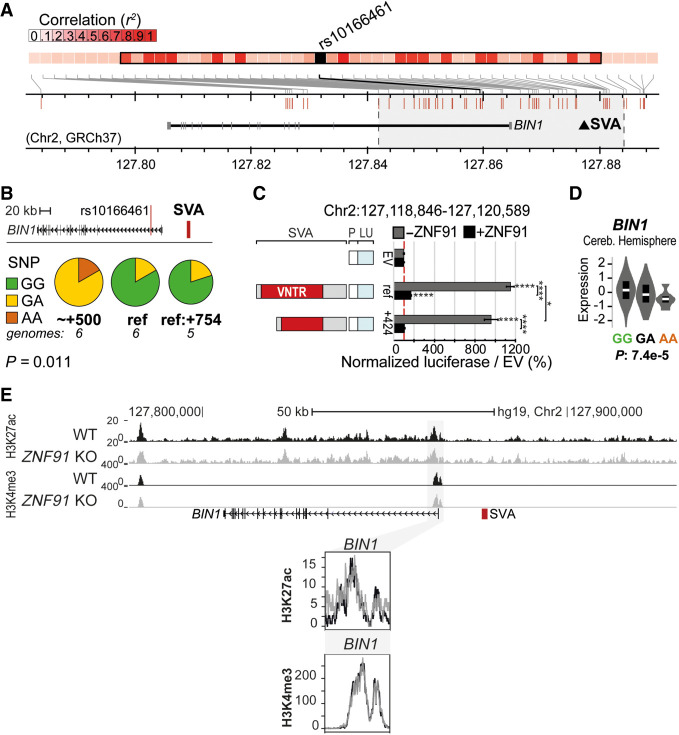
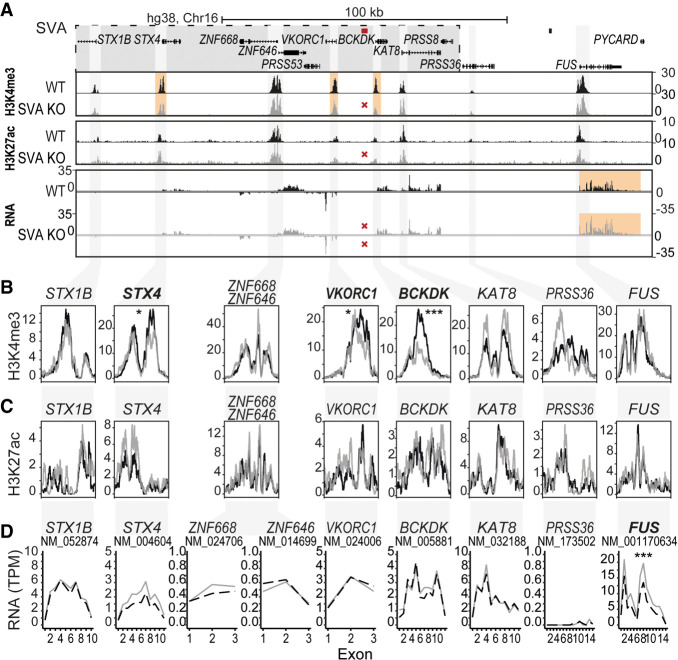
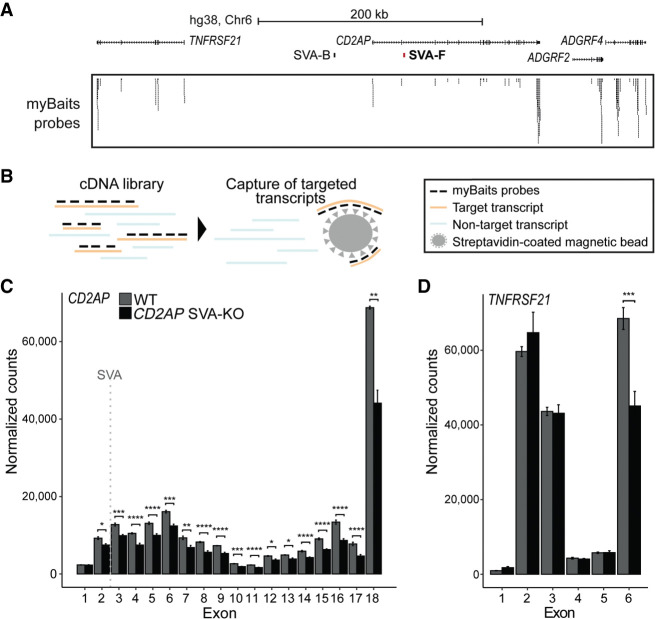
Genome-wide association studies (GWAS) have been highly informative in discovering disease-associated loci but are not designed to capture all structural variations in the human genome. Using long-read sequencing data, we discovered widespread structural variation within SINE-VNTR-Alu (SVA) elements, a class of great ape-specific transposable elements with gene-regulatory roles, which represents a major source of structural variability in the human population. We highlight the presence of structurally variable SVAs (SV-SVAs) in neurological disease-associated loci, and we further associate SV-SVAs to disease-associated SNPs and differential gene expression using luciferase assays and expression quantitative trait loci data. Finally, we genetically deleted SV-SVAs in the BIN1 and CD2AP Alzheimer's disease-associated risk loci and in the BCKDK Parkinson's disease-associated risk locus and assessed multiple aspects of their gene-regulatory influence in a human neuronal context. Together, this study reveals a novel layer of genetic variation in transposable elements that may contribute to identification of the structural variants that are the actual drivers of disease associations of GWAS loci.
© 2022 van Bree et al.; Published by Cold Spring Harbor Laboratory Press.
Figures
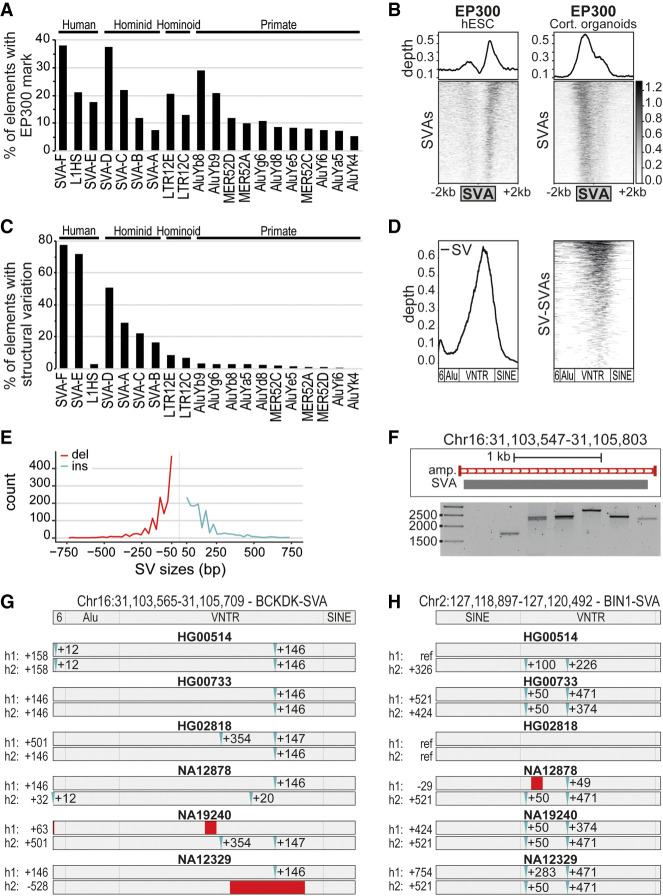
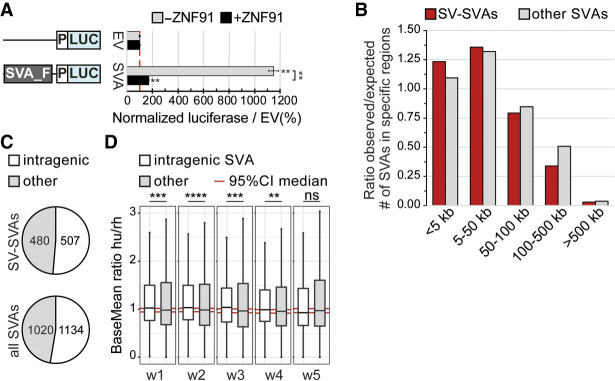
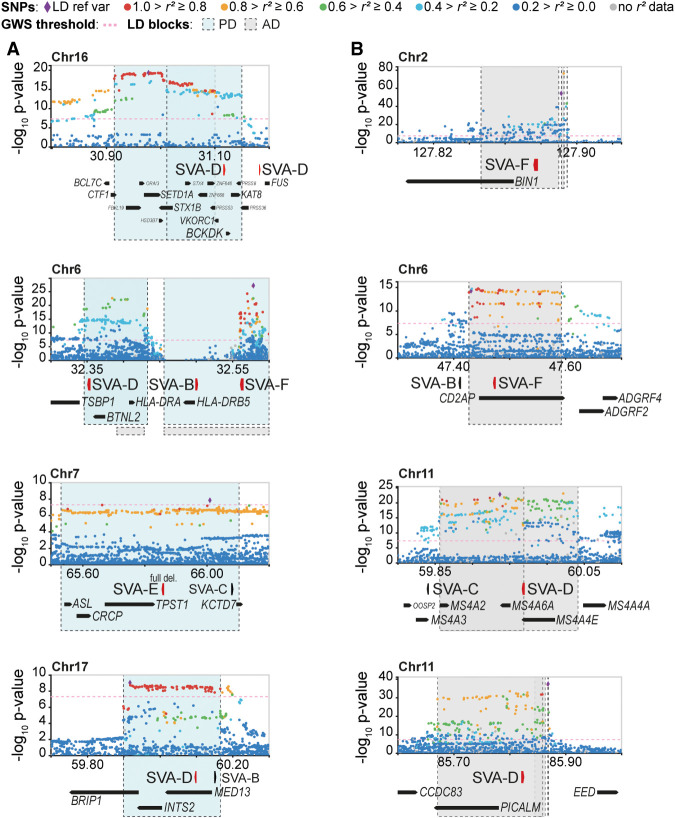
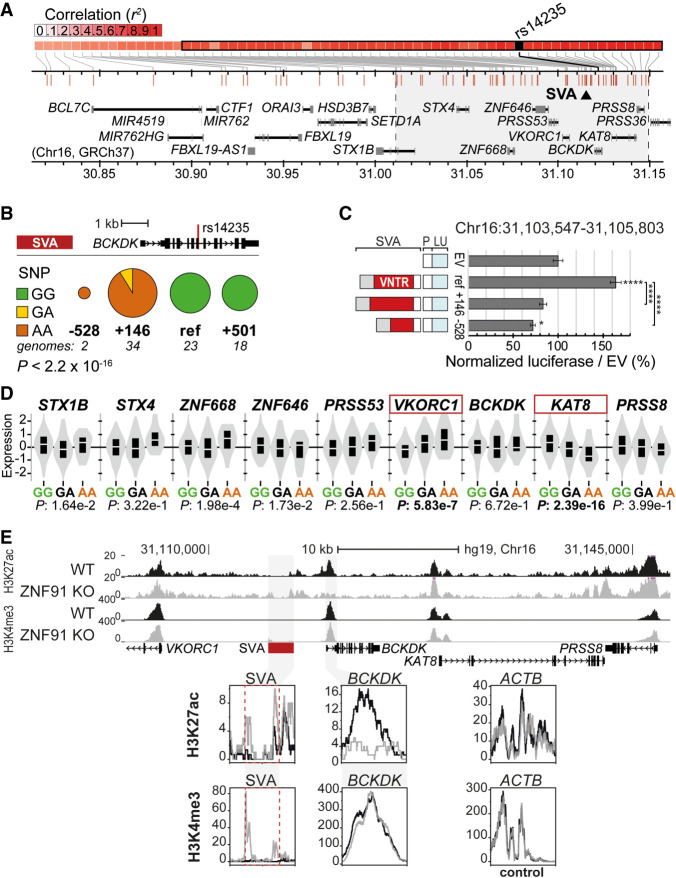
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous