

A single-cell atlas of non-haematopoietic cells in human lymph nodes and lymphoma reveals a landscape of stromal remodelling
- PMID: 35332263
- PMCID: PMC9033586
- DOI: 10.1038/s41556-022-00866-3
A single-cell atlas of non-haematopoietic cells in human lymph nodes and lymphoma reveals a landscape of stromal remodelling
Abstract
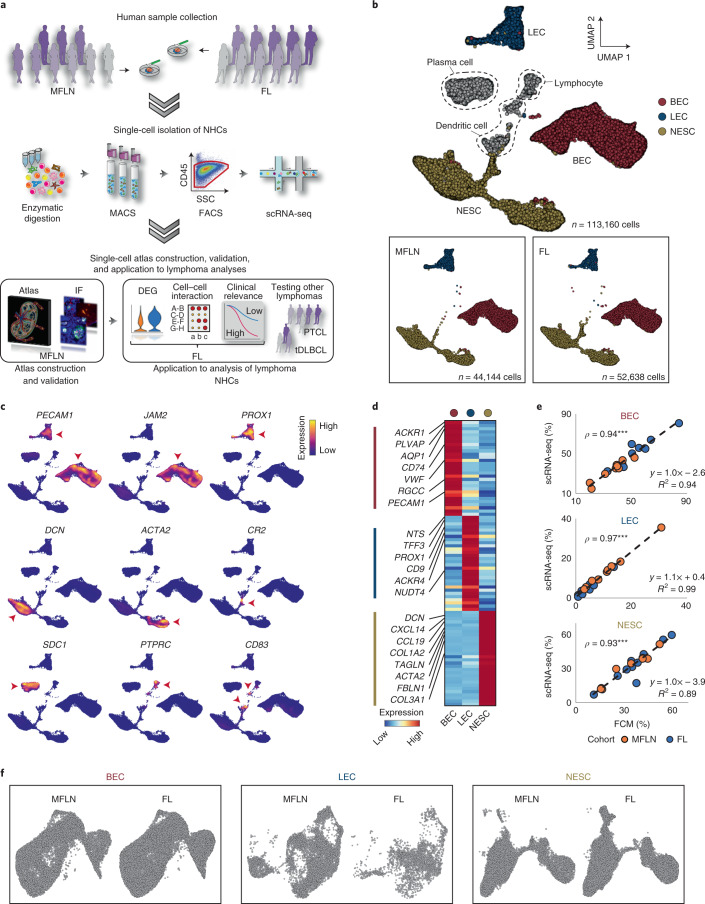
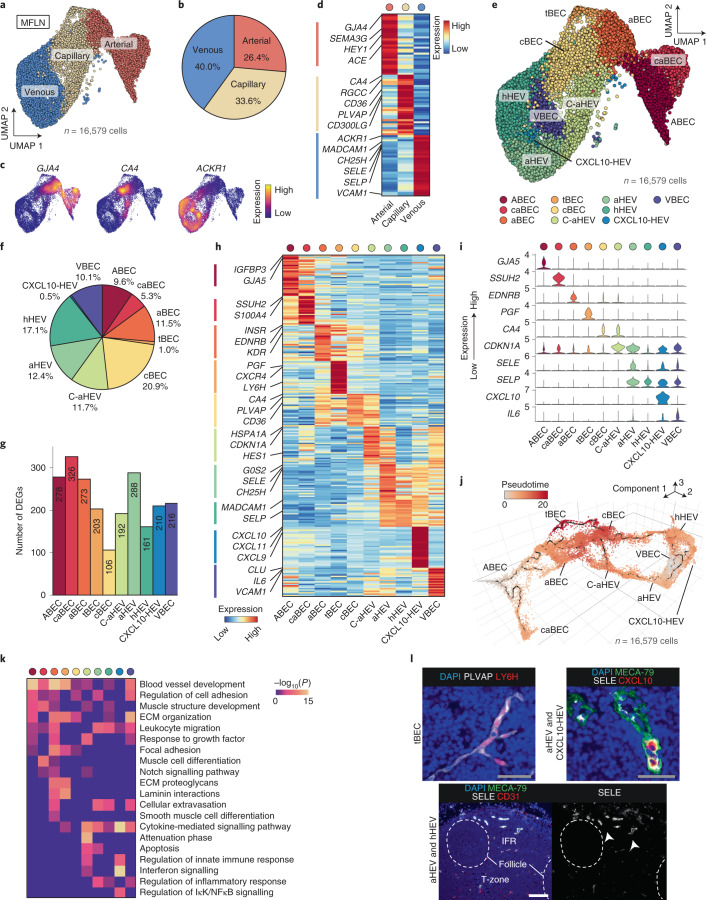
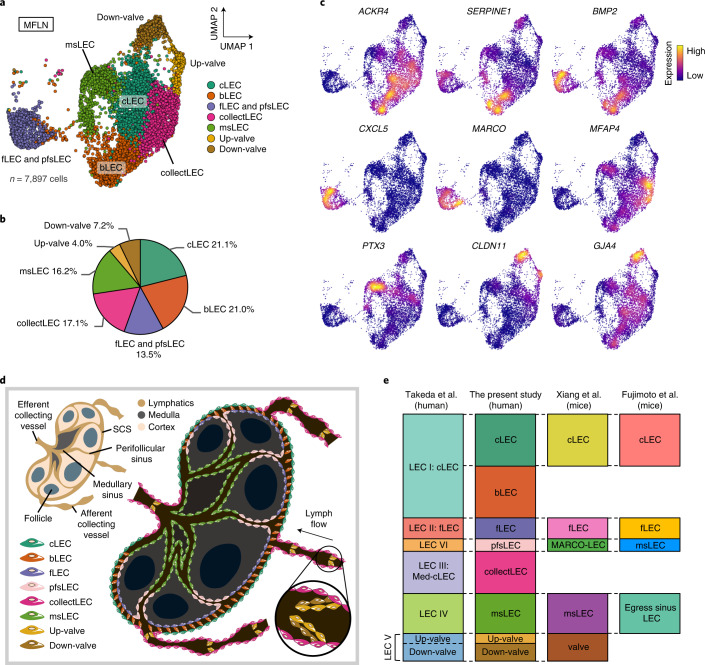
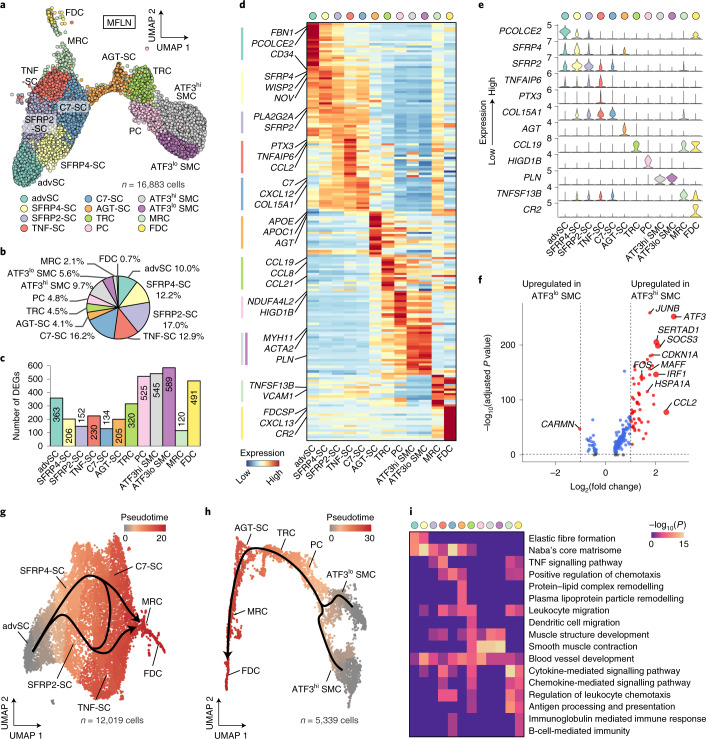
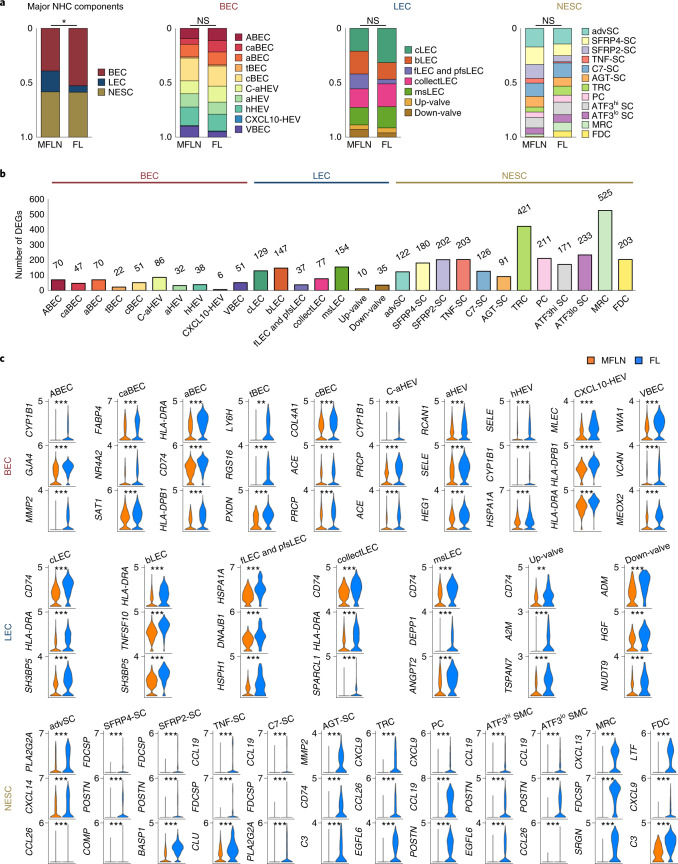
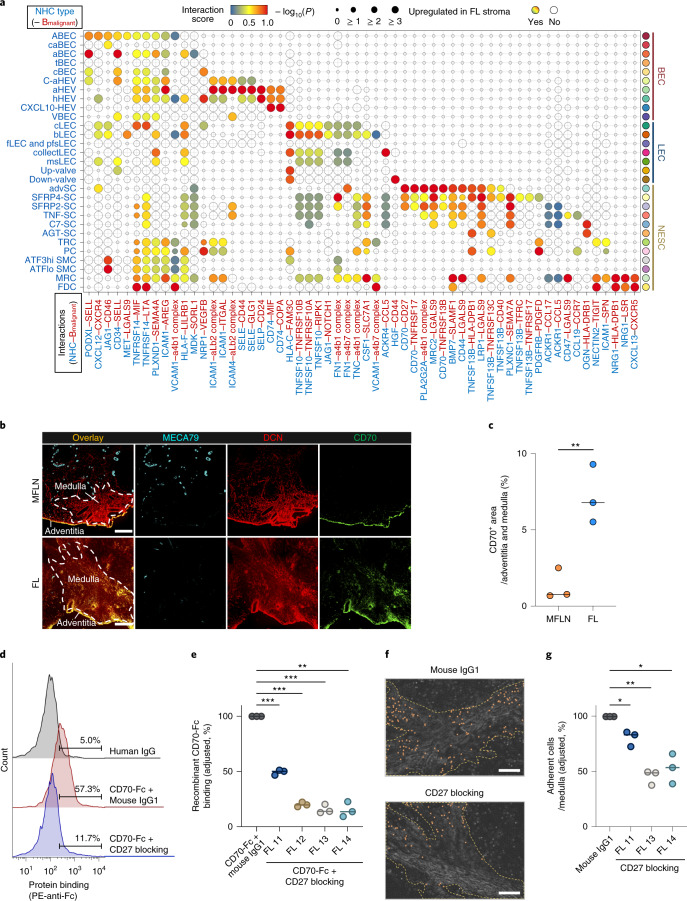
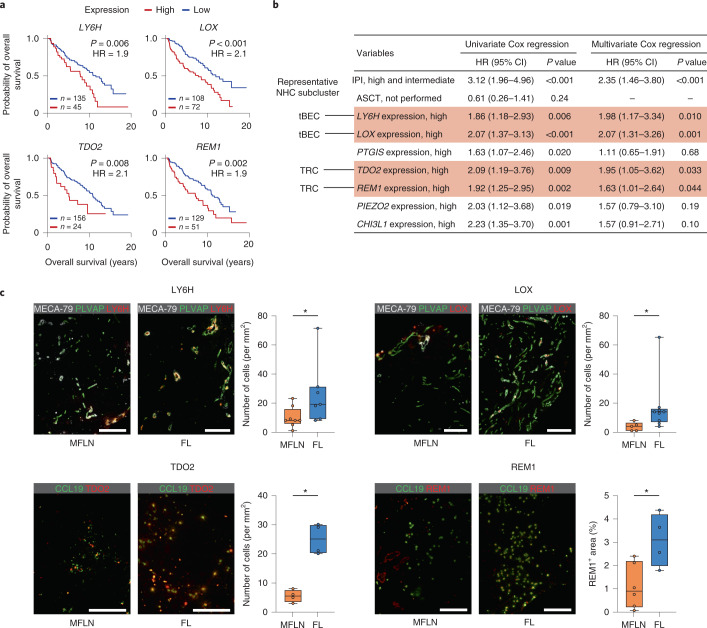
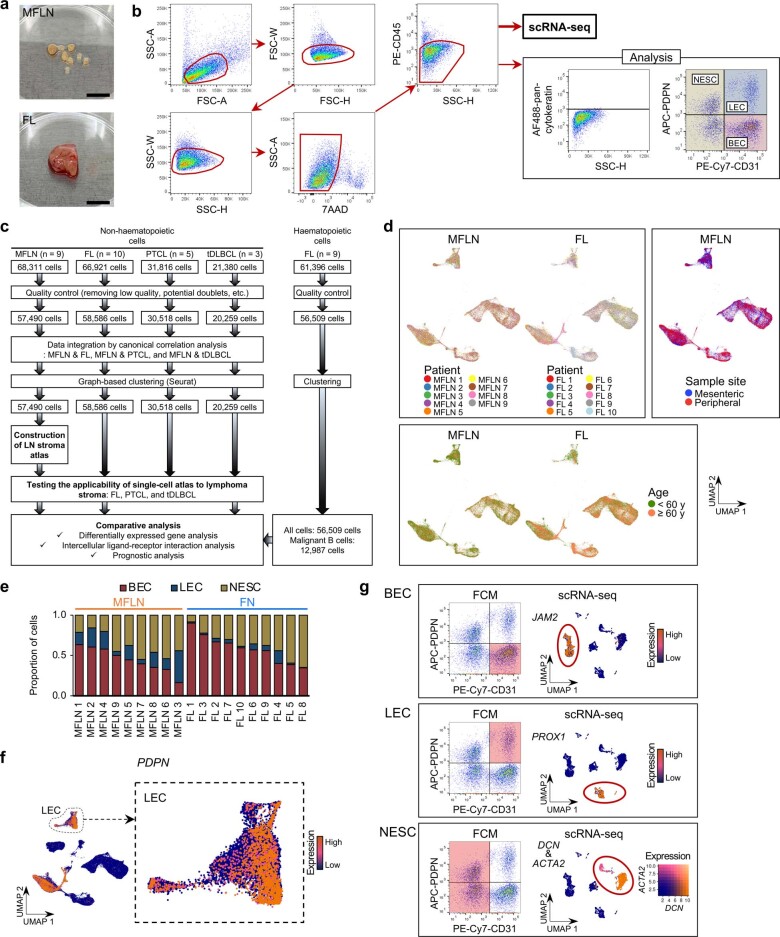
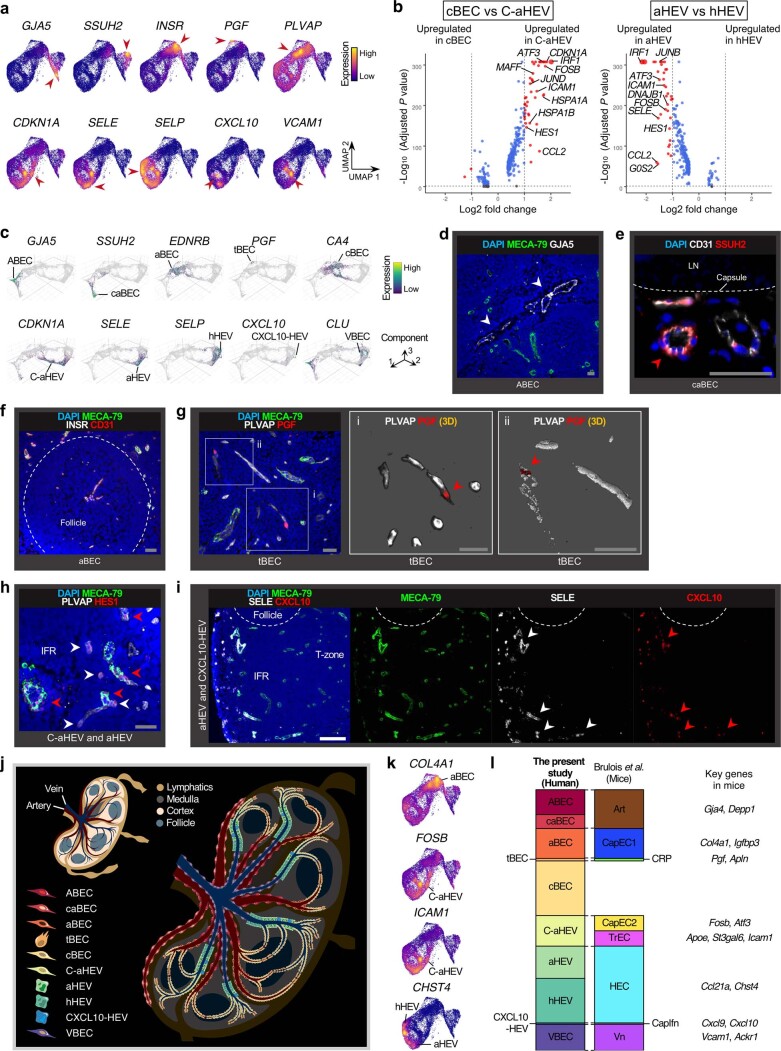
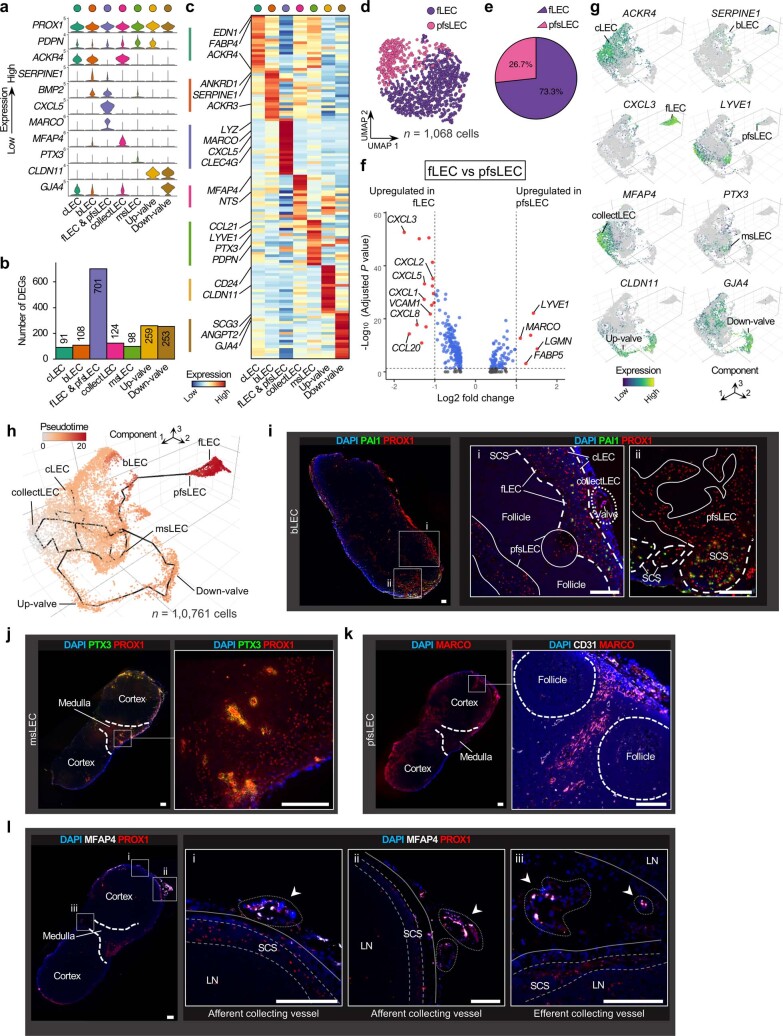
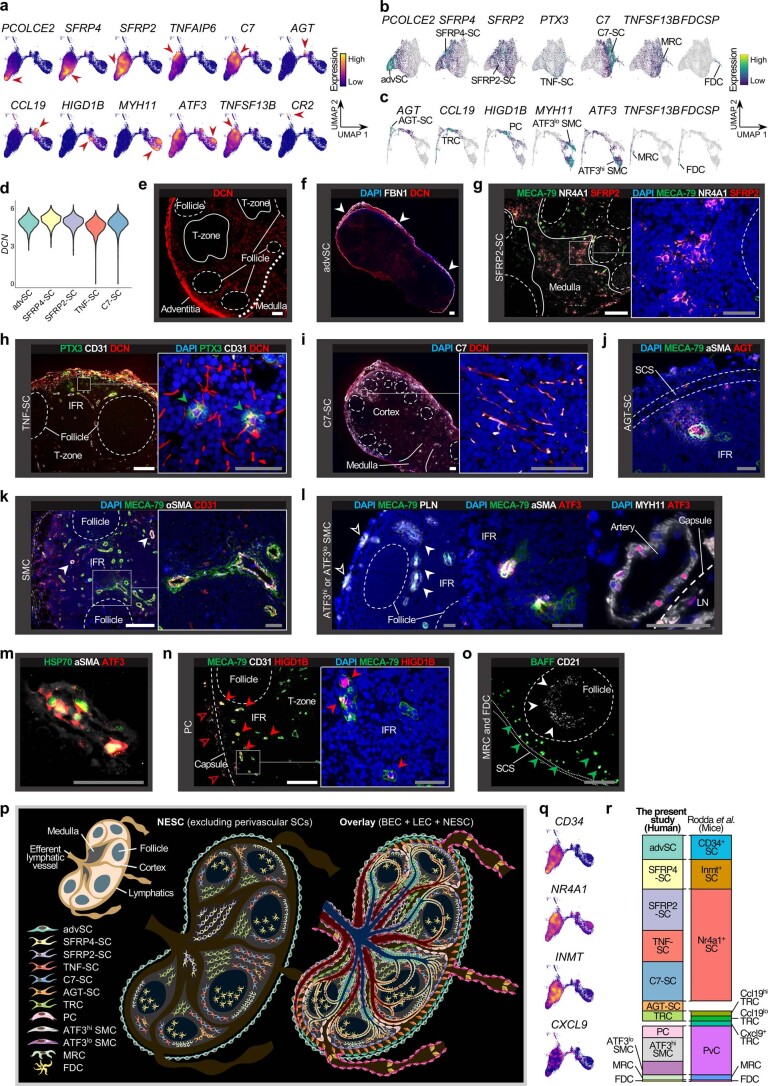
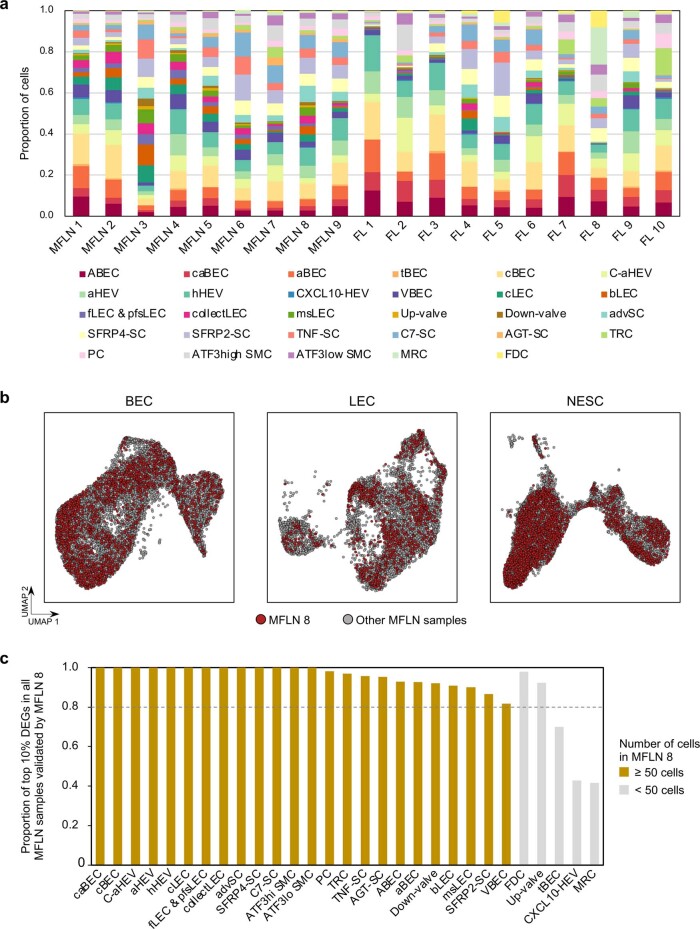
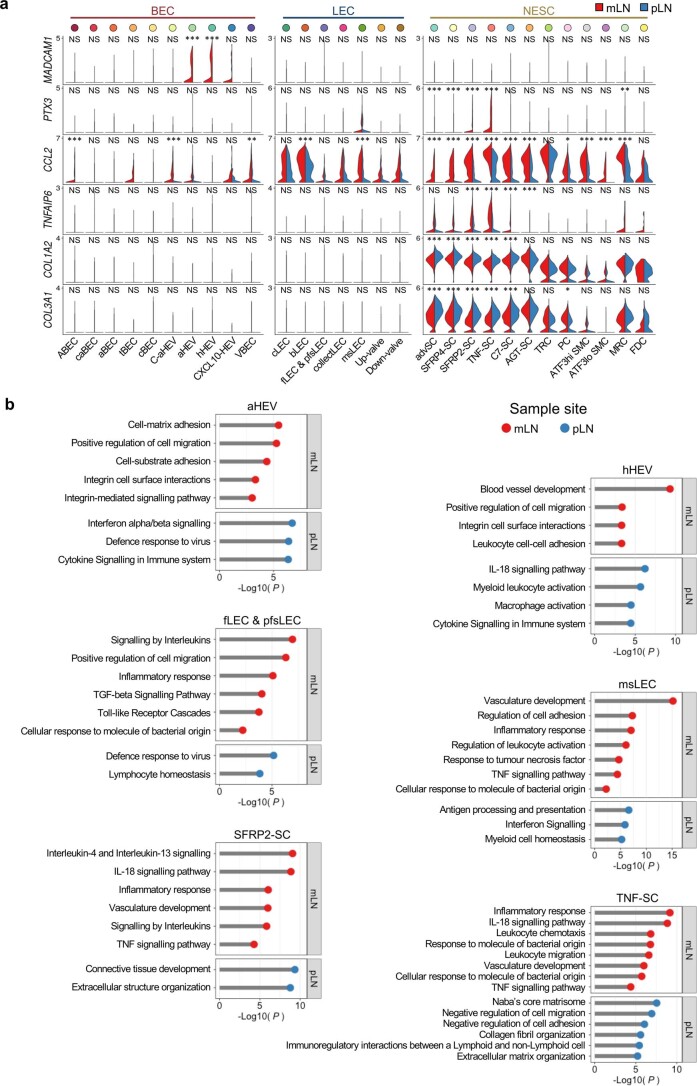
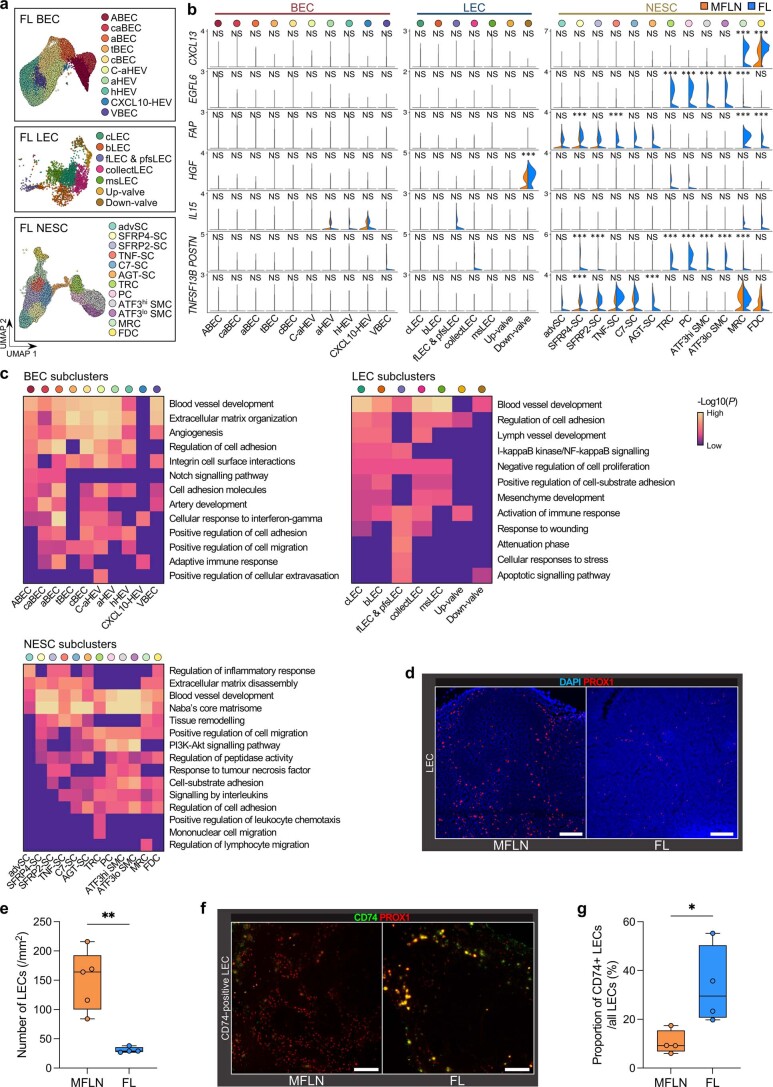
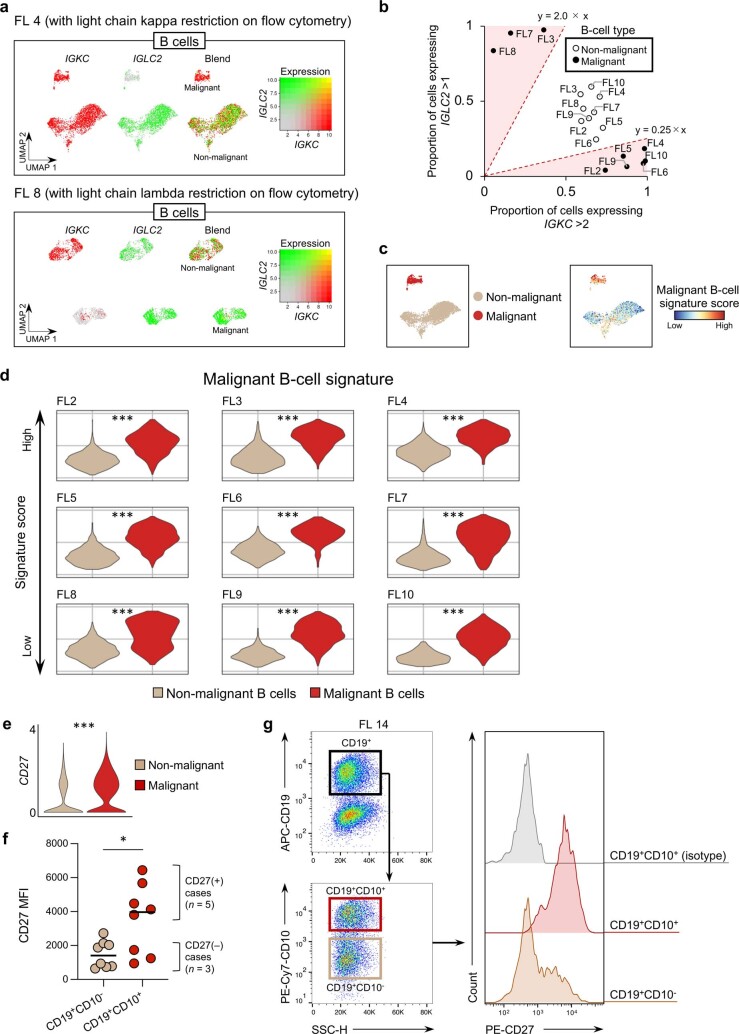
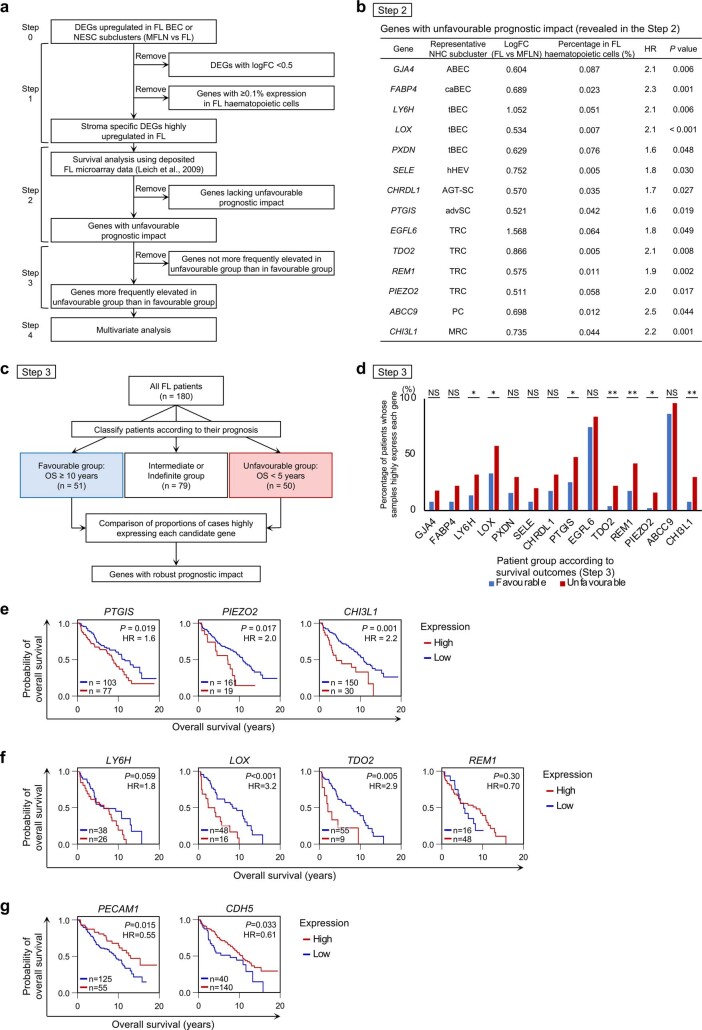
The activities of non-haematopoietic cells (NHCs), including mesenchymal stromal cells and endothelial cells, in lymphomas are reported to underlie lymphomagenesis. However, our understanding of lymphoma NHCs has been hampered by unexplained NHC heterogeneity, even in normal human lymph nodes (LNs). Here we constructed a single-cell transcriptome atlas of more than 100,000 NHCs collected from 27 human samples, including LNs and various nodal lymphomas, and it revealed 30 distinct subclusters, including some that were previously unrecognized. Notably, this atlas was useful for comparative analyses with lymphoma NHCs, which revealed an unanticipated landscape of subcluster-specific changes in gene expression and interaction with malignant cells in follicular lymphoma NHCs. This facilitates our understanding of stromal remodelling in lymphoma and highlights potential clinical biomarkers. Our study largely updates NHC taxonomy in human LNs and analysis of disease status, and provides a rich resource and deeper insights into LN and lymphoma biology to advance lymphoma management and therapy.
© 2022. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
References
-
- Swerdlow, S. et al. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues 4th edn (WHO Press, 2017).
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Medical
