

Design of protein-binding proteins from the target structure alone
- PMID: 35332283
- PMCID: PMC9117152
- DOI: 10.1038/s41586-022-04654-9
Design of protein-binding proteins from the target structure alone
Abstract
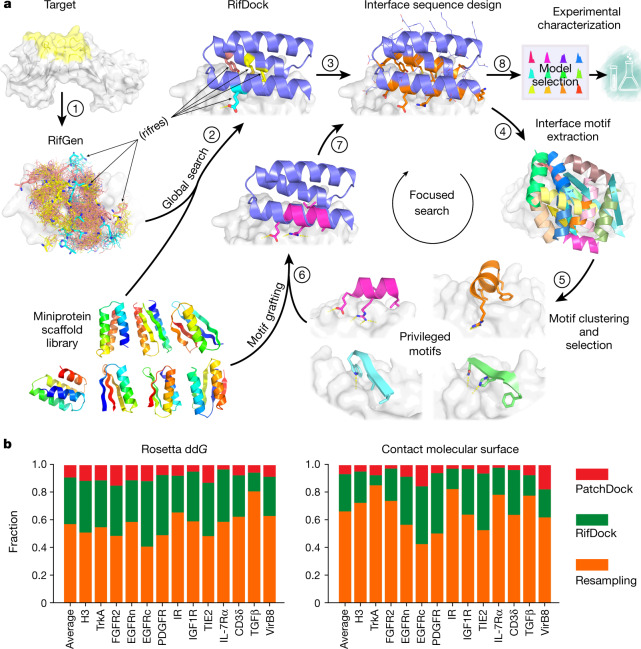
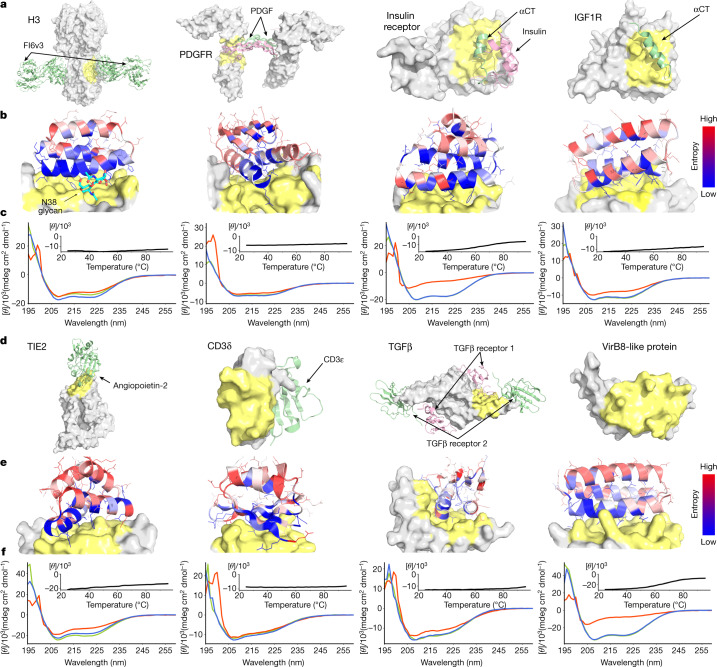
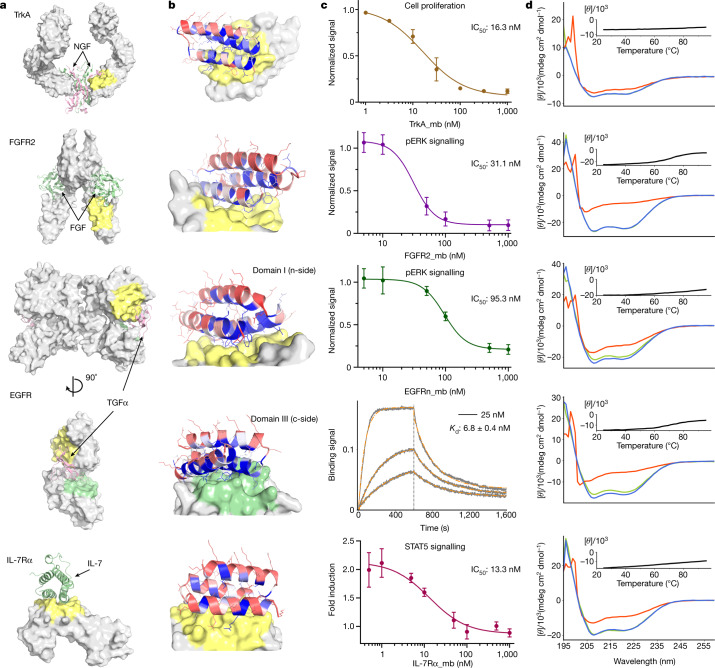
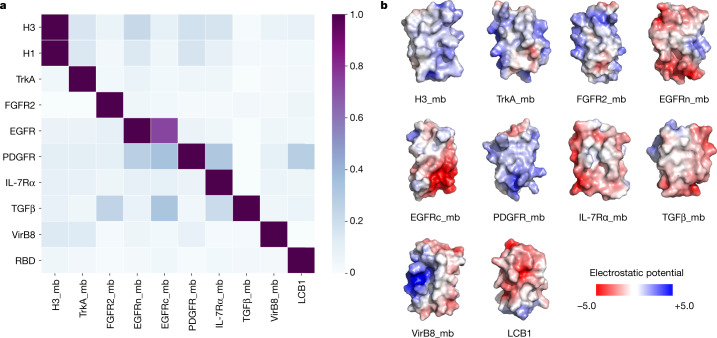
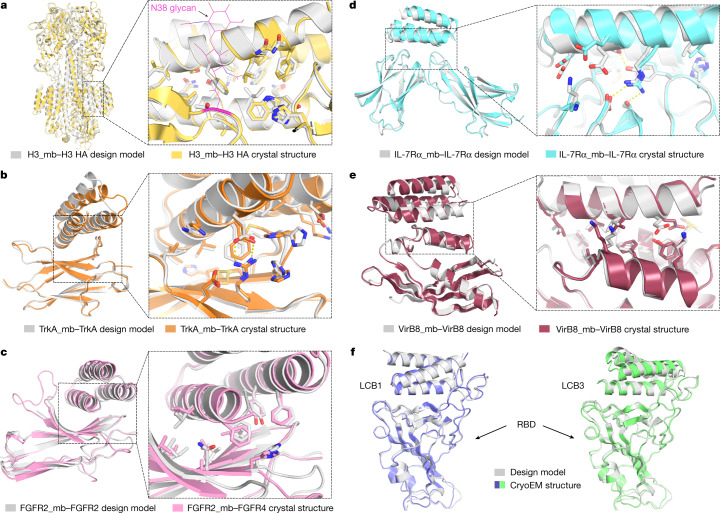
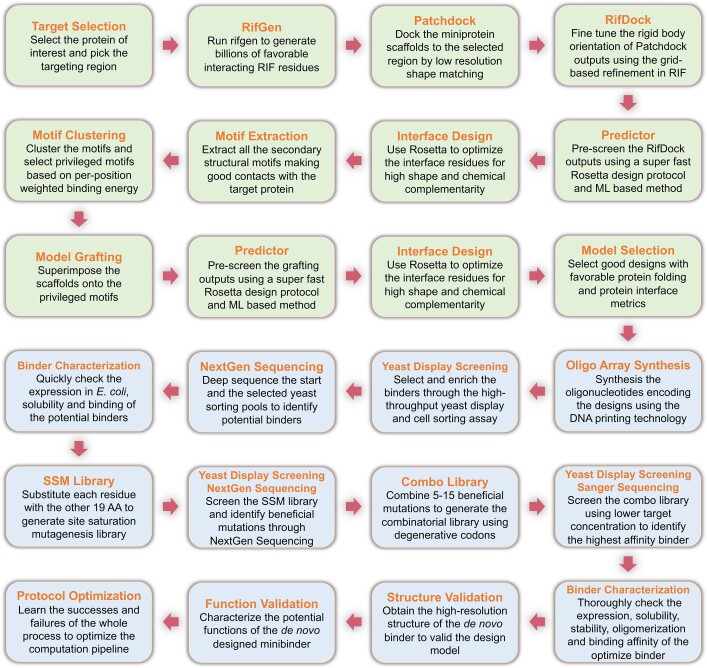
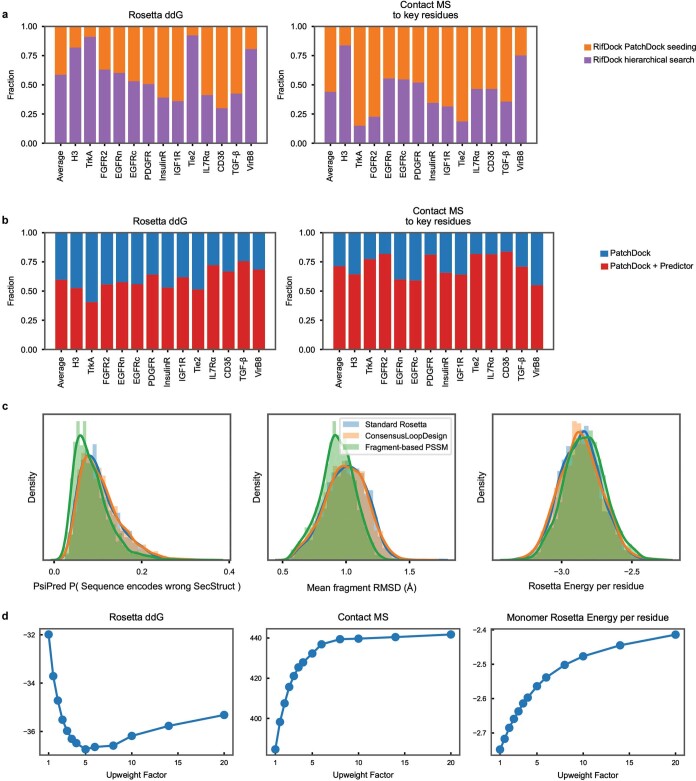
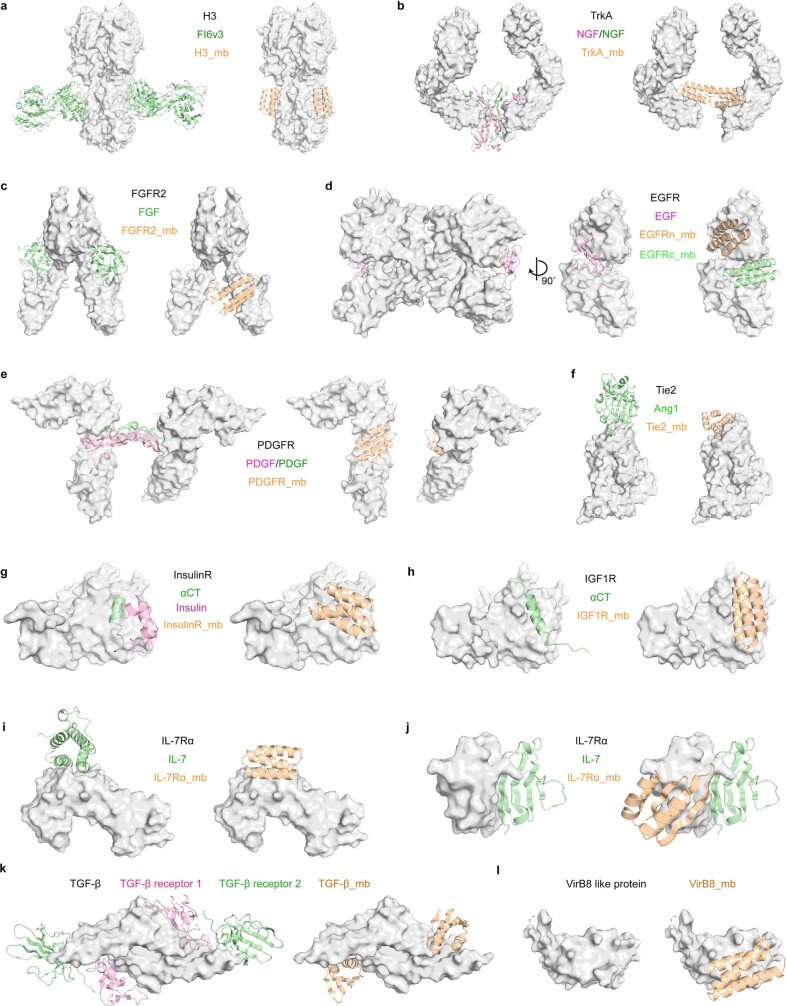
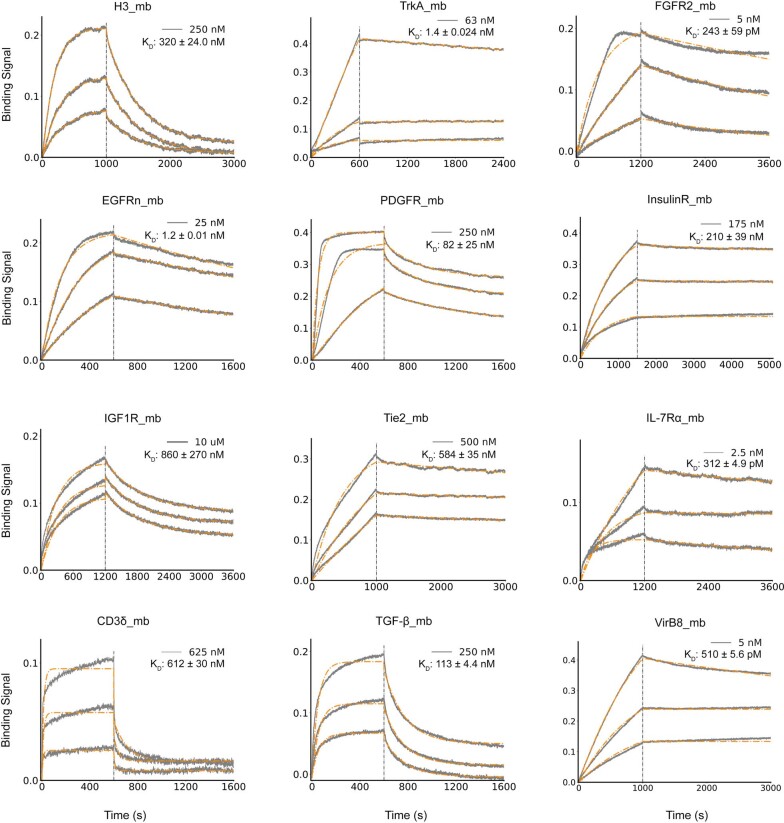
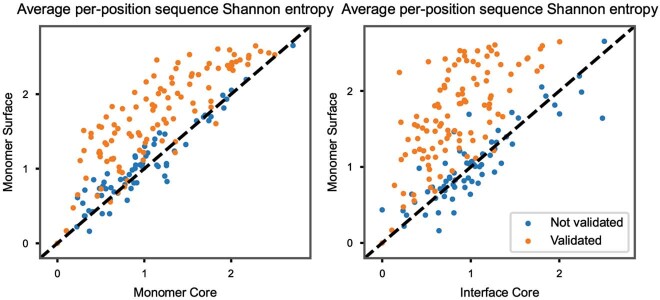
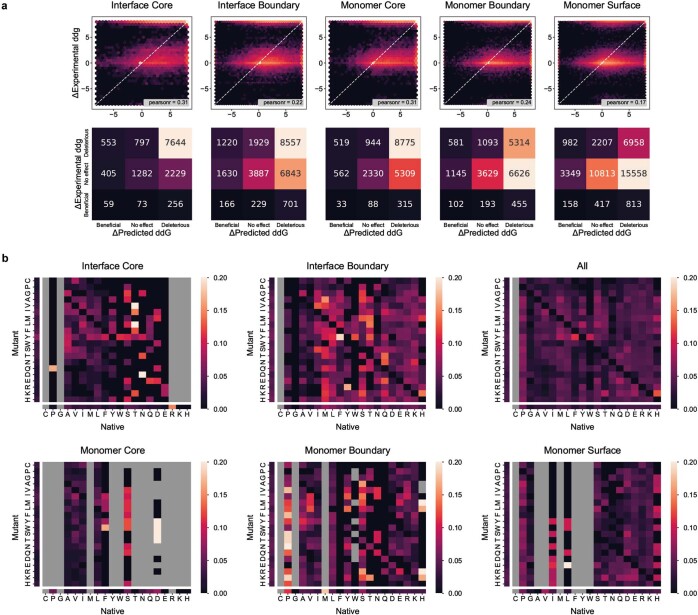
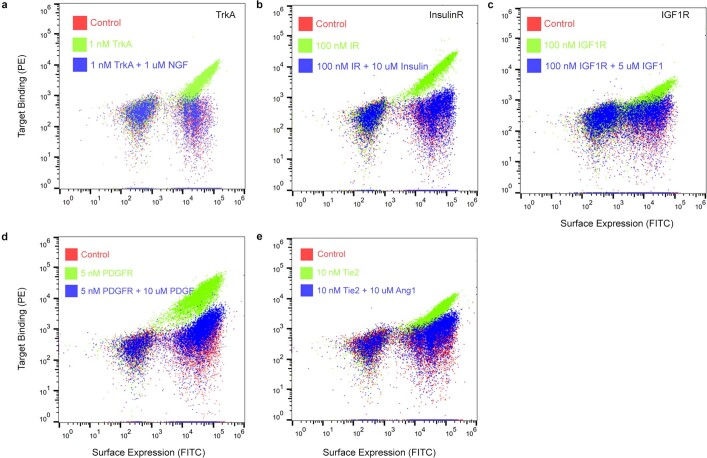
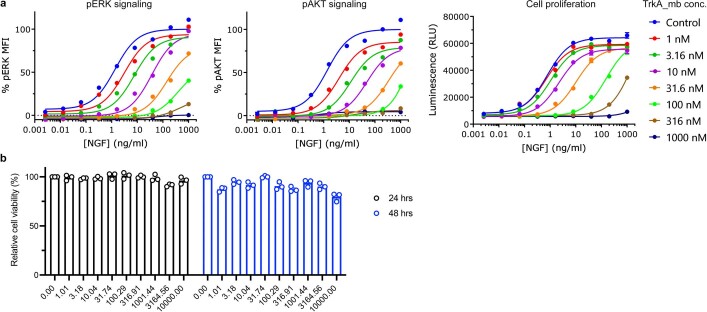
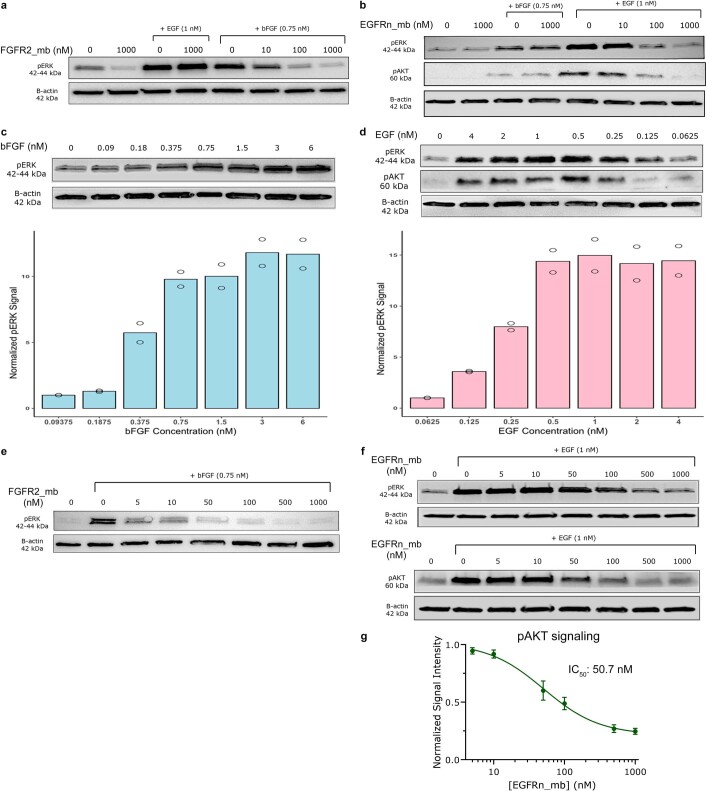
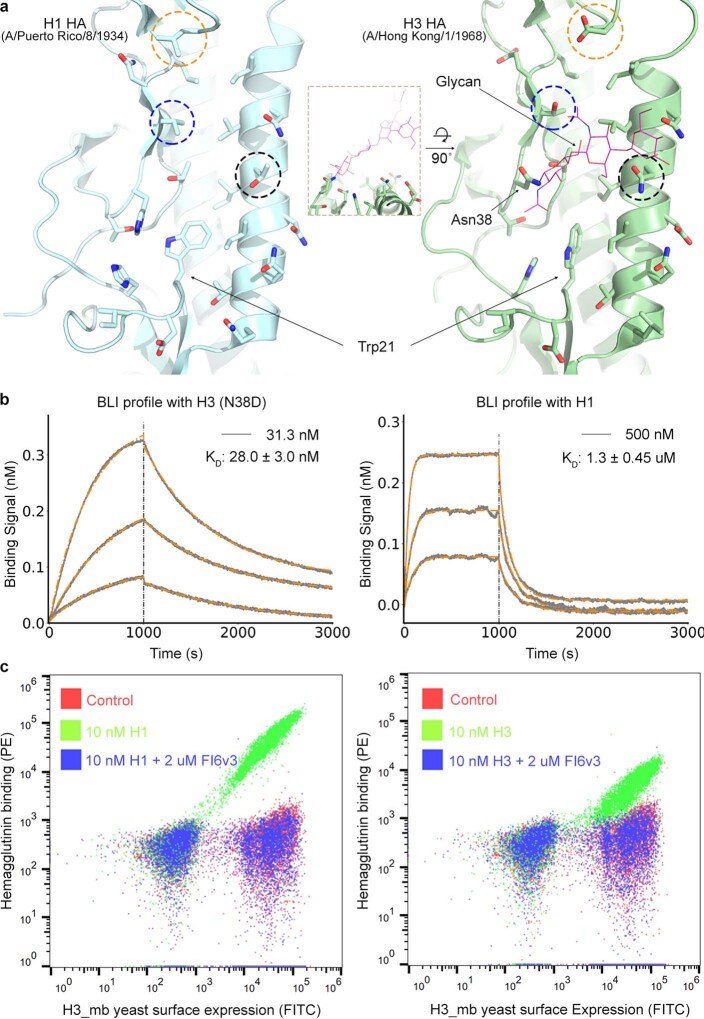
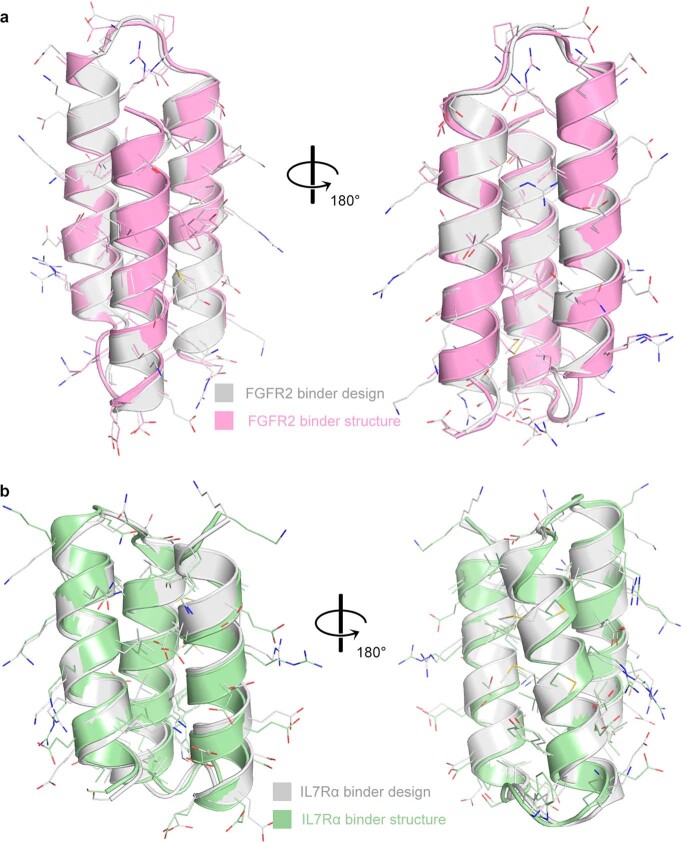
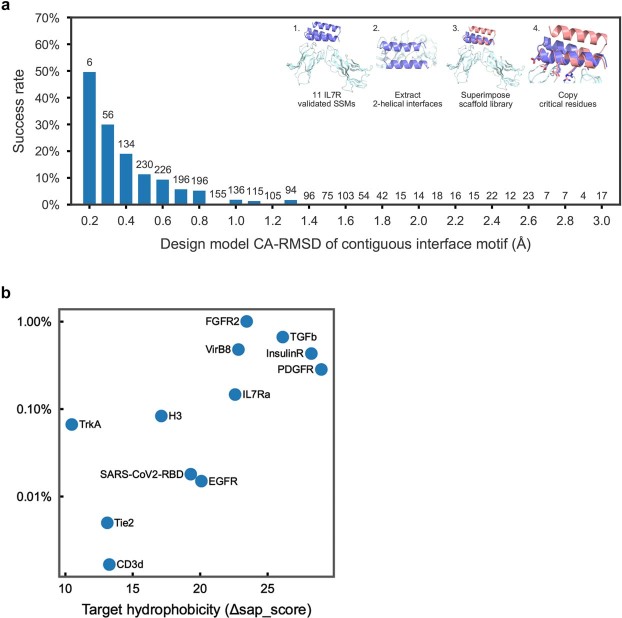
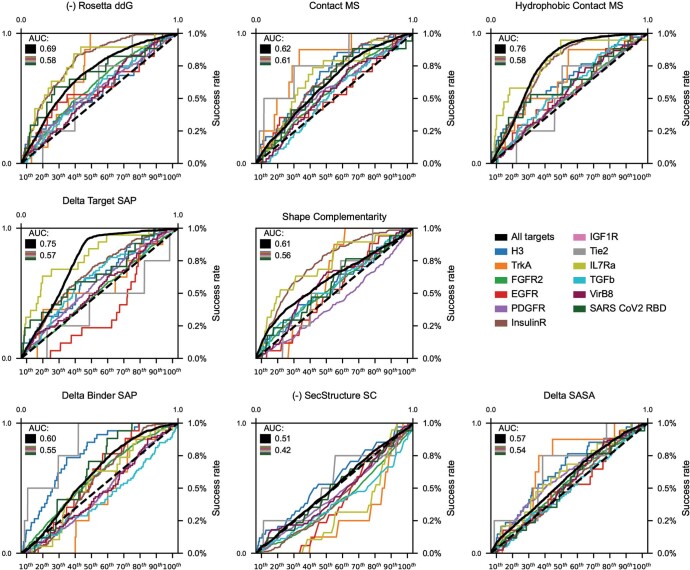
The design of proteins that bind to a specific site on the surface of a target protein using no information other than the three-dimensional structure of the target remains a challenge1-5. Here we describe a general solution to this problem that starts with a broad exploration of the vast space of possible binding modes to a selected region of a protein surface, and then intensifies the search in the vicinity of the most promising binding modes. We demonstrate the broad applicability of this approach through the de novo design of binding proteins to 12 diverse protein targets with different shapes and surface properties. Biophysical characterization shows that the binders, which are all smaller than 65 amino acids, are hyperstable and, following experimental optimization, bind their targets with nanomolar to picomolar affinities. We succeeded in solving crystal structures of five of the binder-target complexes, and all five closely match the corresponding computational design models. Experimental data on nearly half a million computational designs and hundreds of thousands of point mutants provide detailed feedback on the strengths and limitations of the method and of our current understanding of protein-protein interactions, and should guide improvements of both. Our approach enables the targeted design of binders to sites of interest on a wide variety of proteins for therapeutic and diagnostic applications.
© 2022. This is a U.S. government work and not under copyright protection in the U.S.; foreign copyright protection may apply.
Conflict of interest statement
L.C., B.C., I.G., B.H., N.B., E.-M.S., L.S. and D.B. are co-inventors on a provisional patent application (21-0753-US-PRO) that incorporates discoveries described in this manuscript.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
- R01 AI140245/AI/NIAID NIH HHS/United States
- R37 AI051321/AI/NIAID NIH HHS/United States
- S10 OD012289/OD/NIH HHS/United States
- P30 GM138396/GM/NIGMS NIH HHS/United States
- HHSN272201700059C/AI/NIAID NIH HHS/United States
- P30 GM124165/GM/NIGMS NIH HHS/United States
- R01 AI150855/AI/NIAID NIH HHS/United States
- P41 GM103393/GM/NIGMS NIH HHS/United States
- HHMI/Howard Hughes Medical Institute/United States
- R01 AG063845/AG/NIA NIH HHS/United States
- R01 AI150885/AI/NIAID NIH HHS/United States
- S10 OD021527/OD/NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
