

HLA autoimmune risk alleles restrict the hypervariable region of T cell receptors
- PMID: 35332318
- PMCID: PMC9010379
- DOI: 10.1038/s41588-022-01032-z
HLA autoimmune risk alleles restrict the hypervariable region of T cell receptors
Abstract
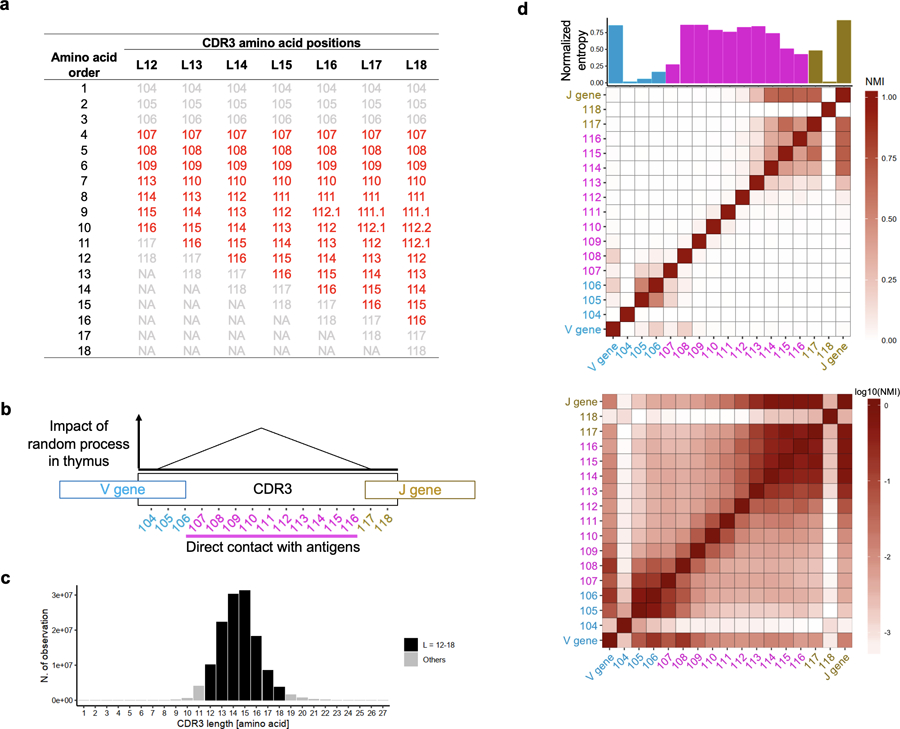
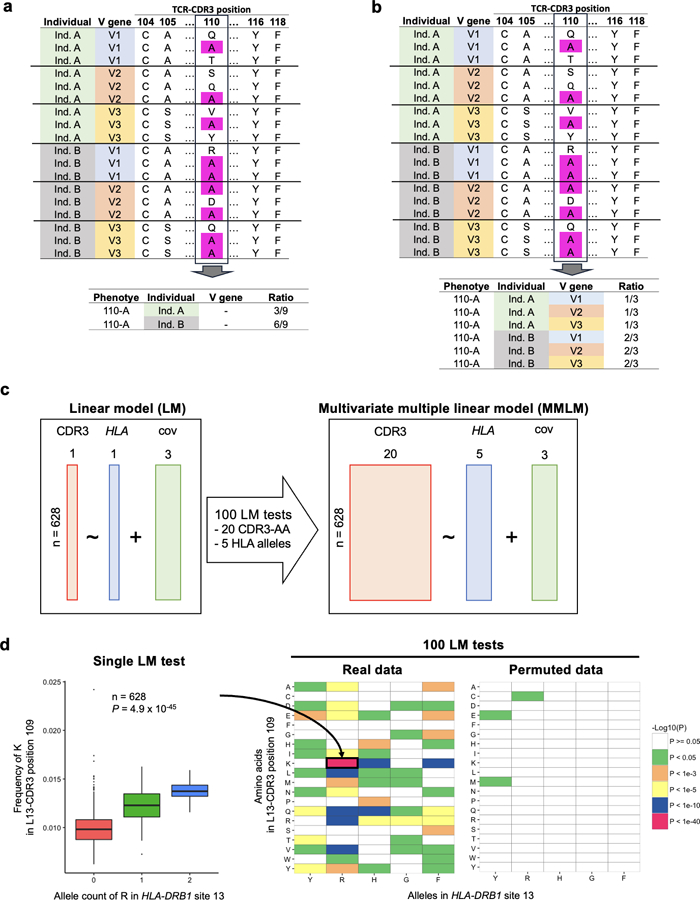
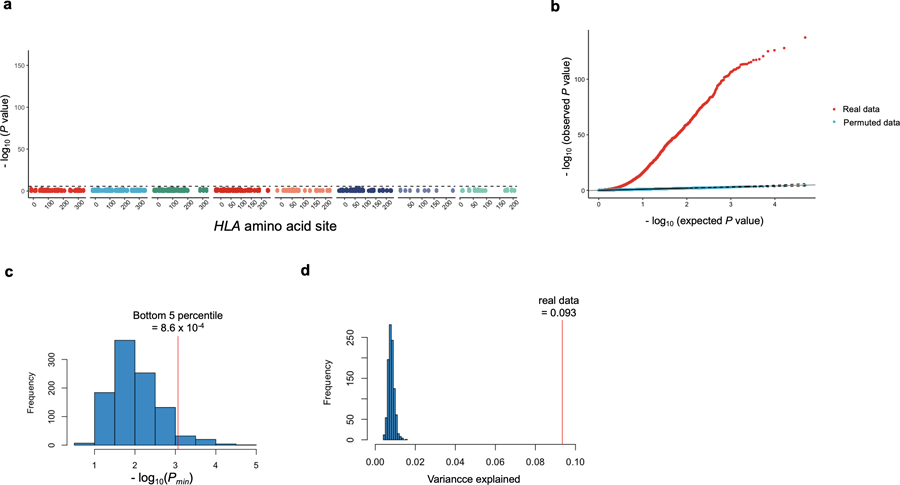
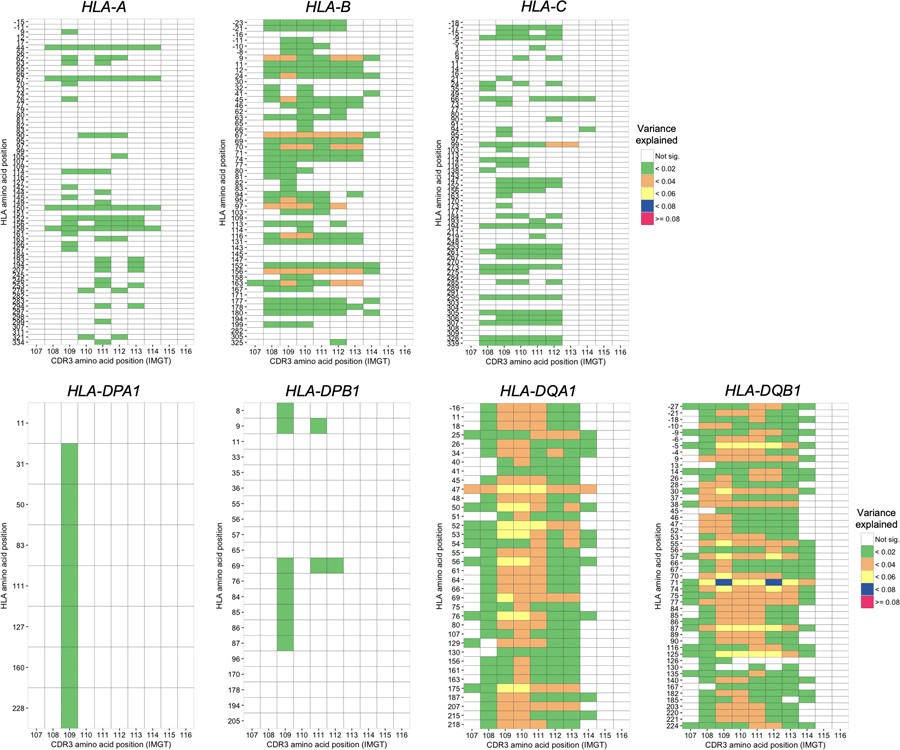
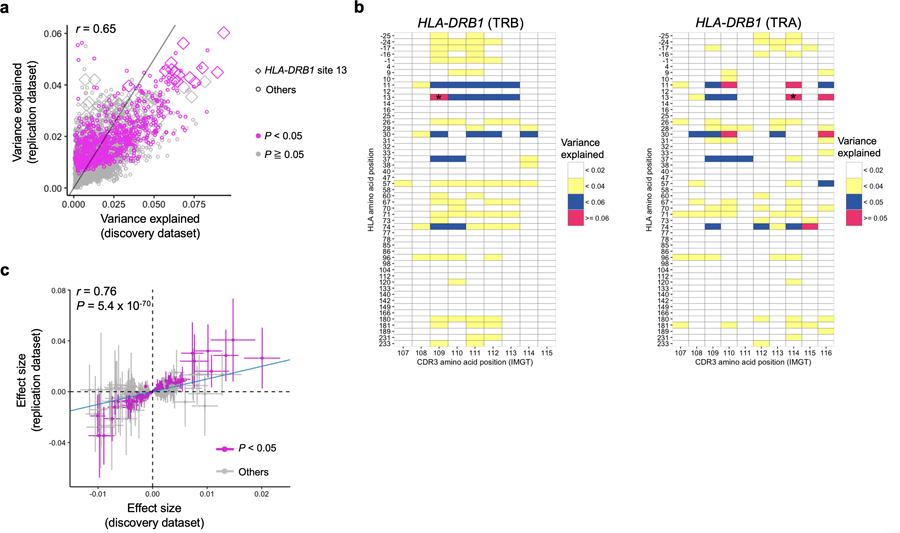
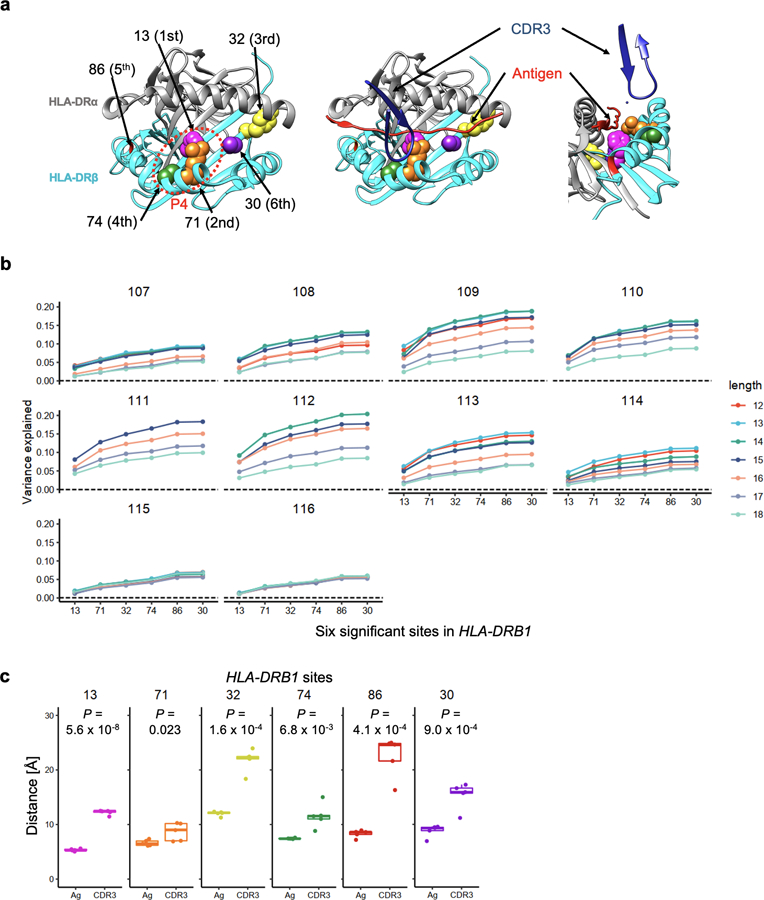
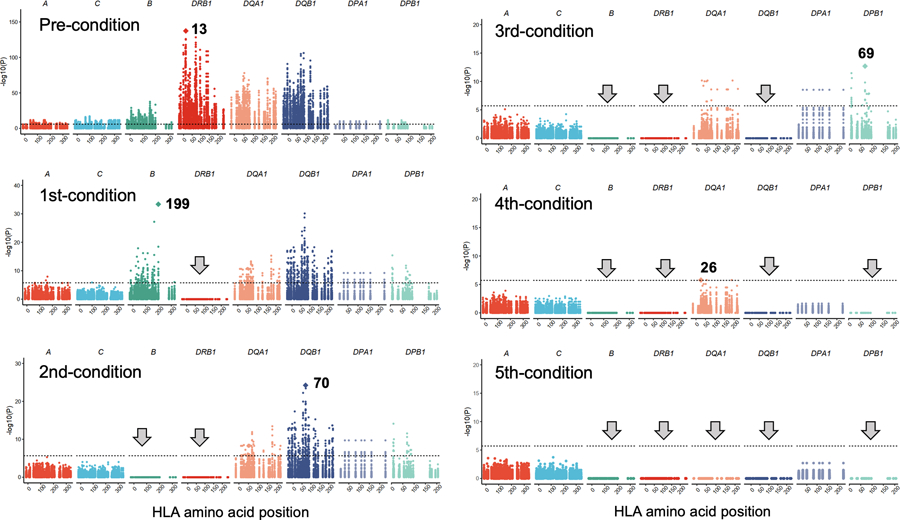
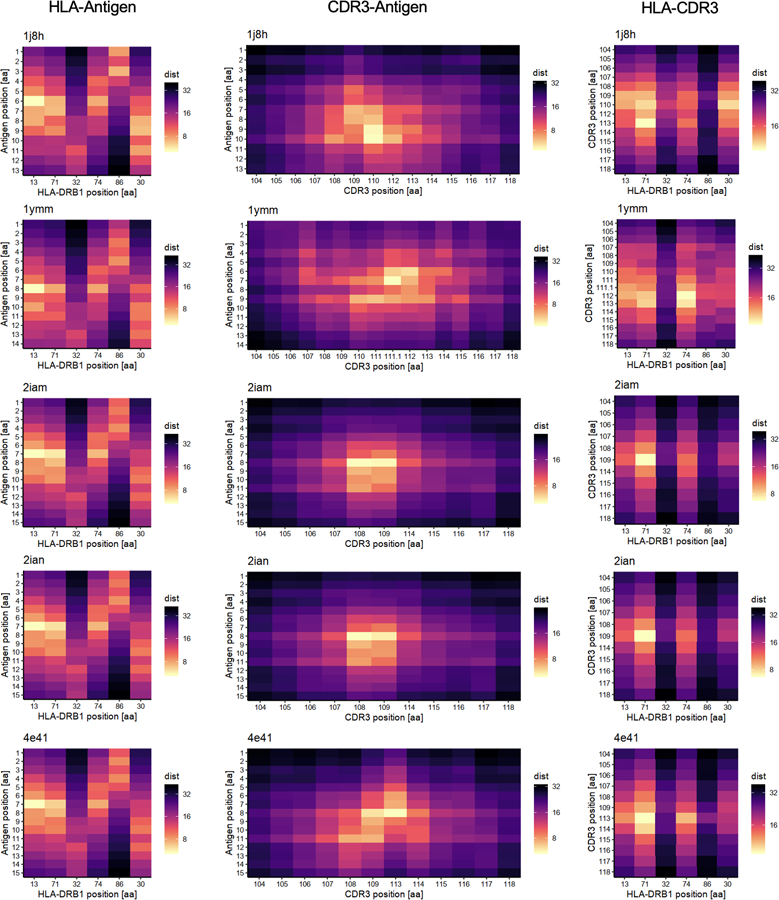
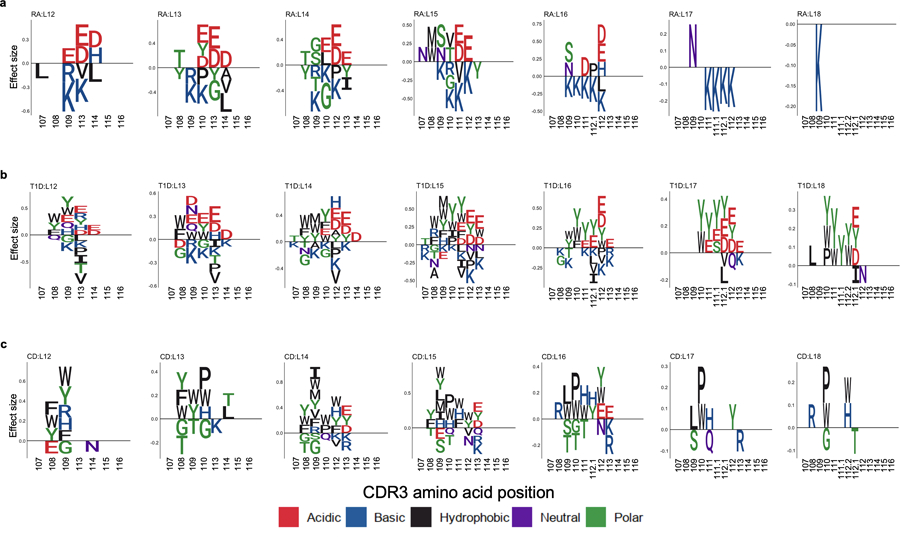
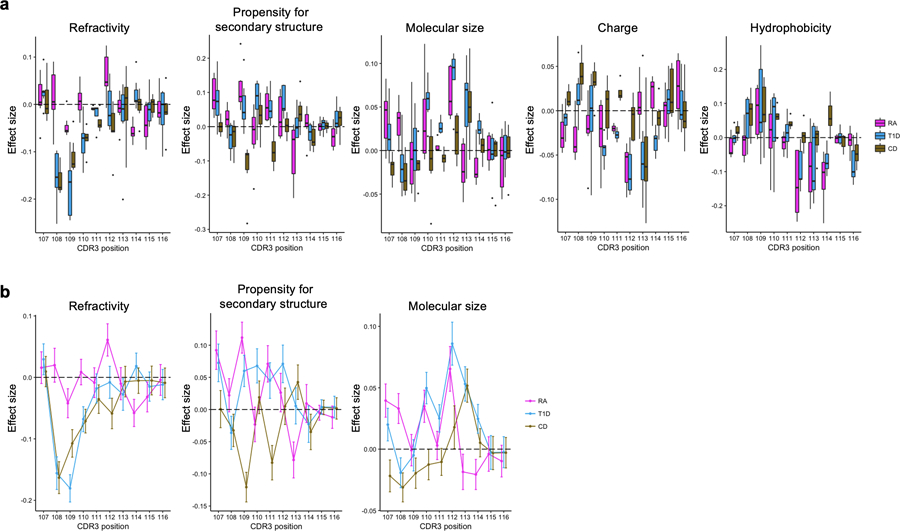
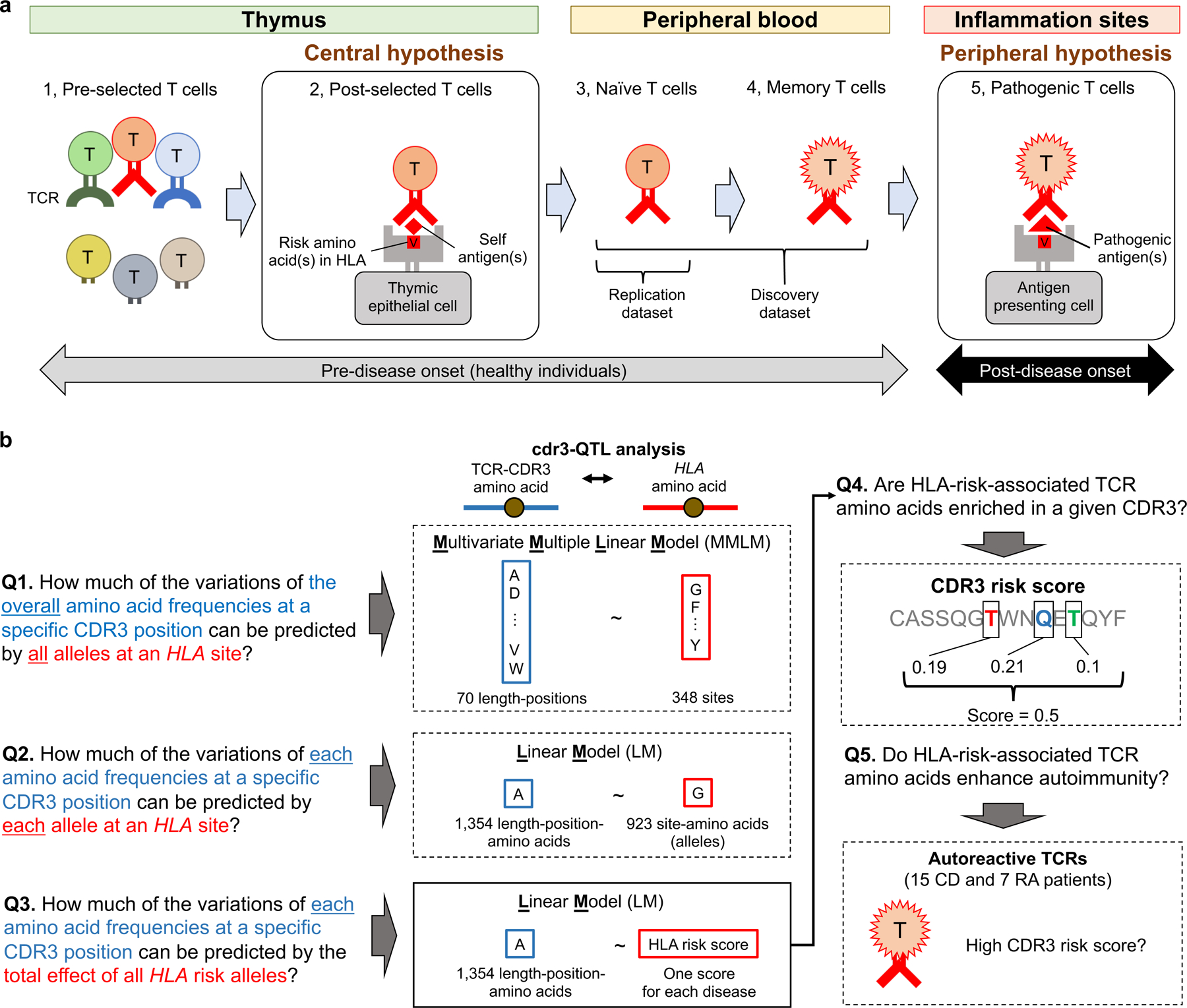
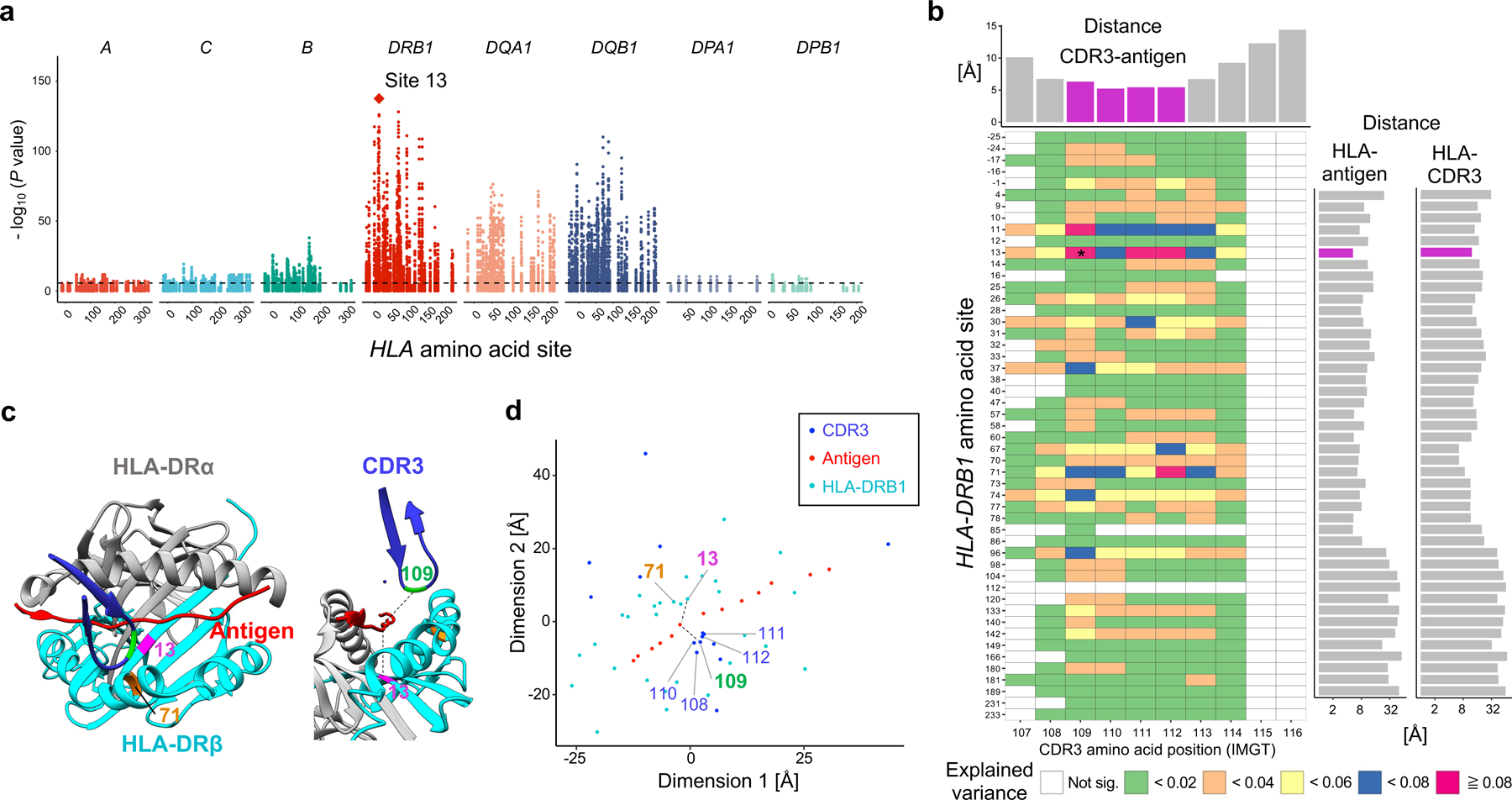
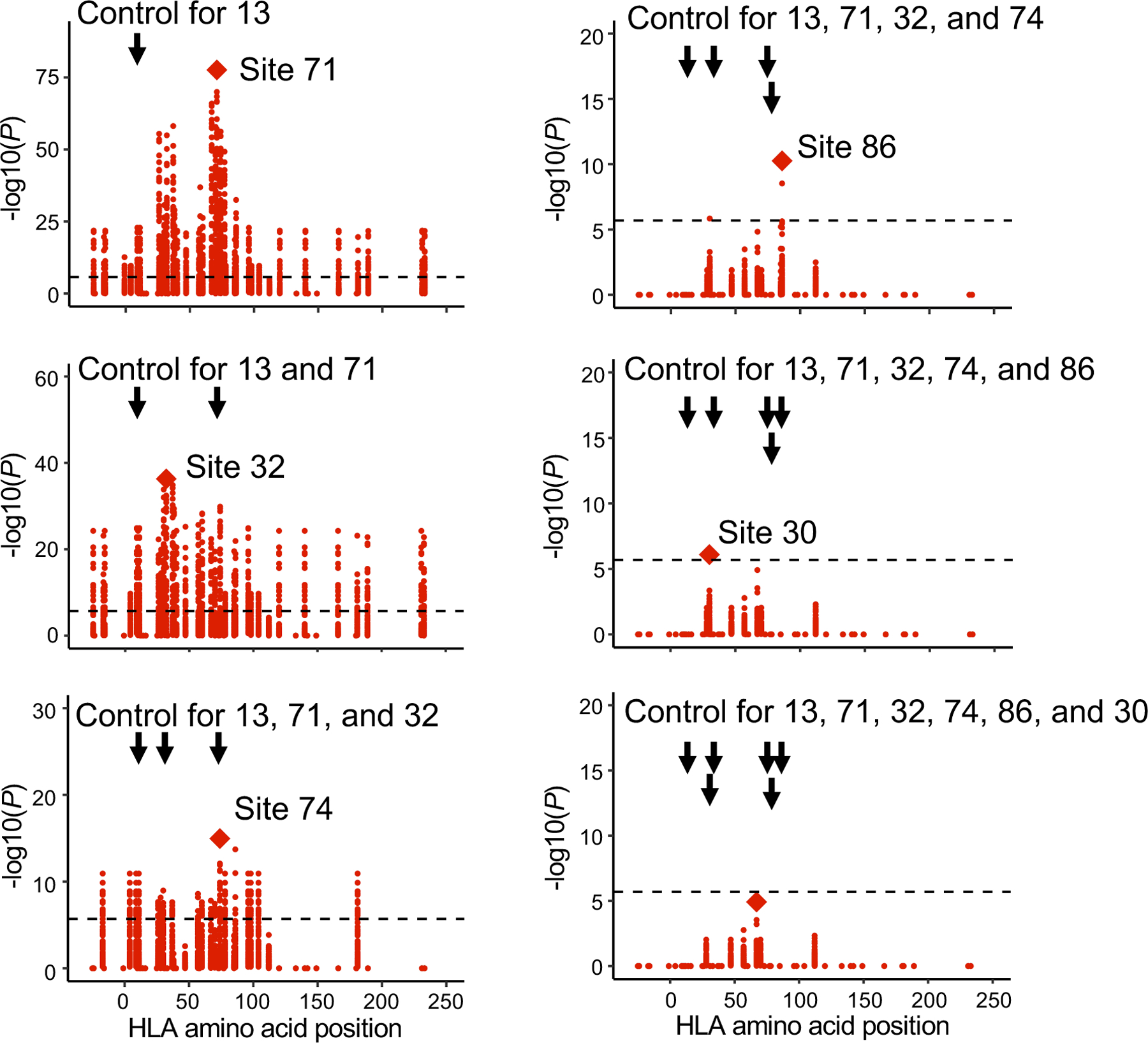
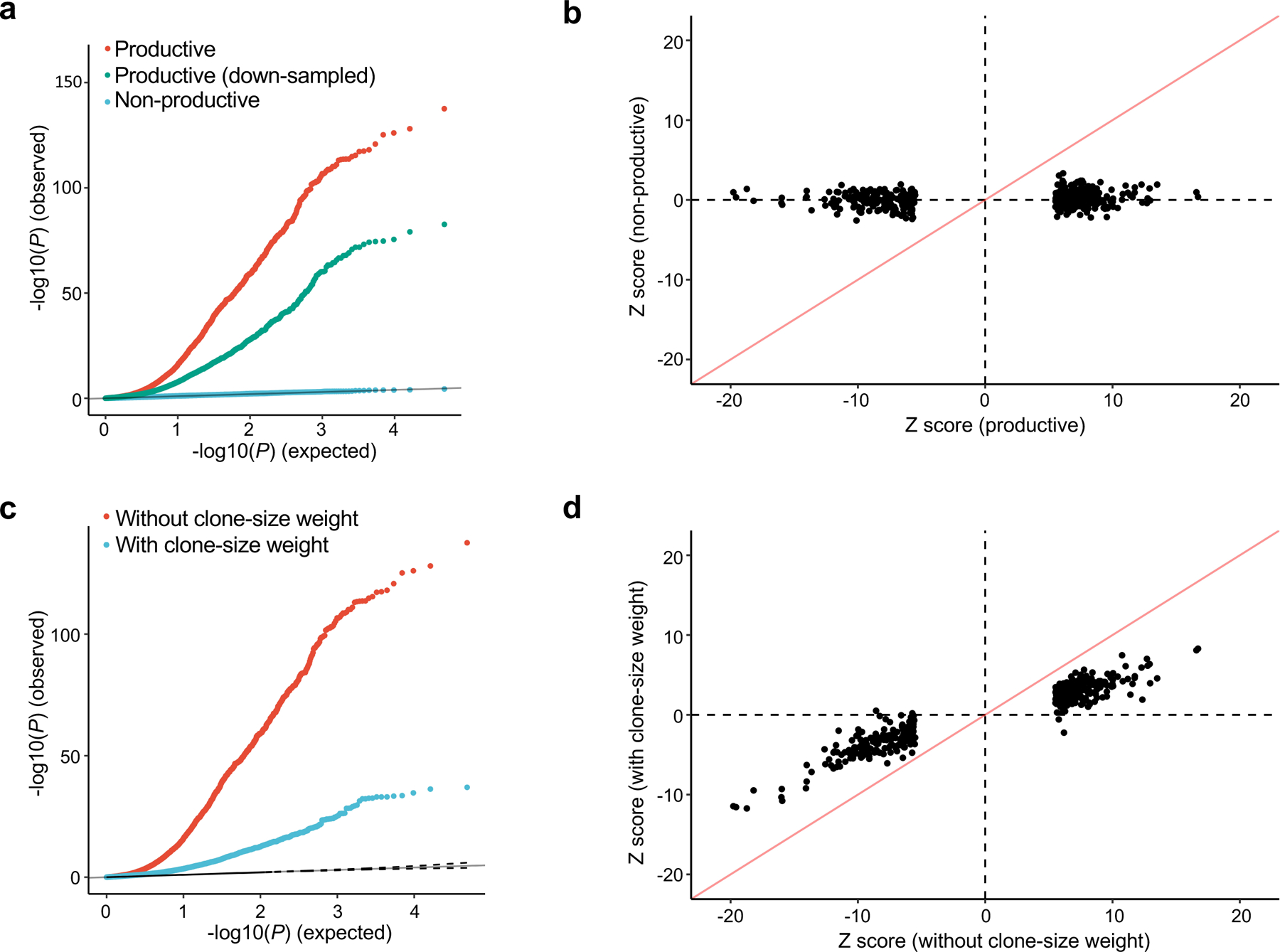
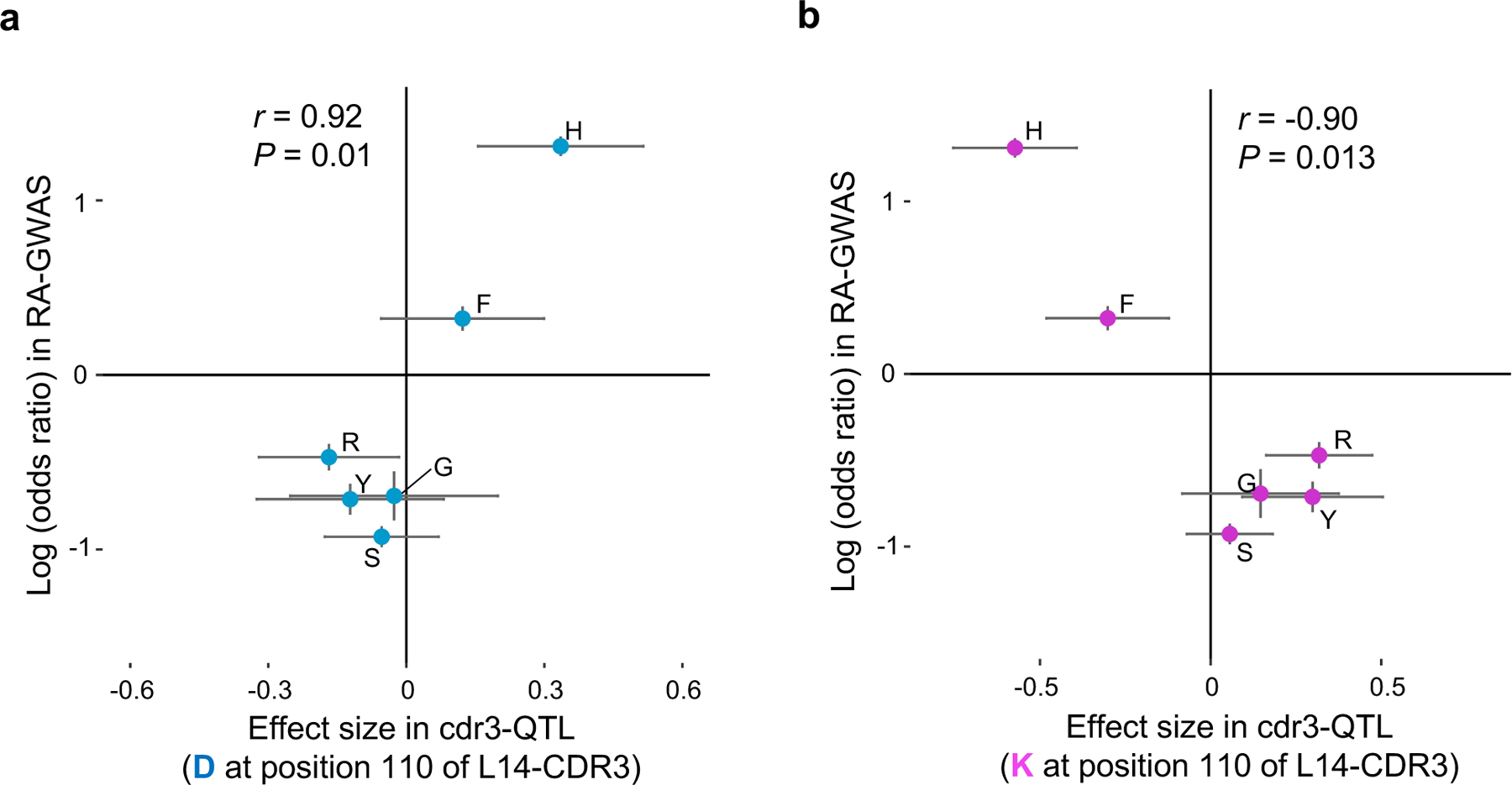
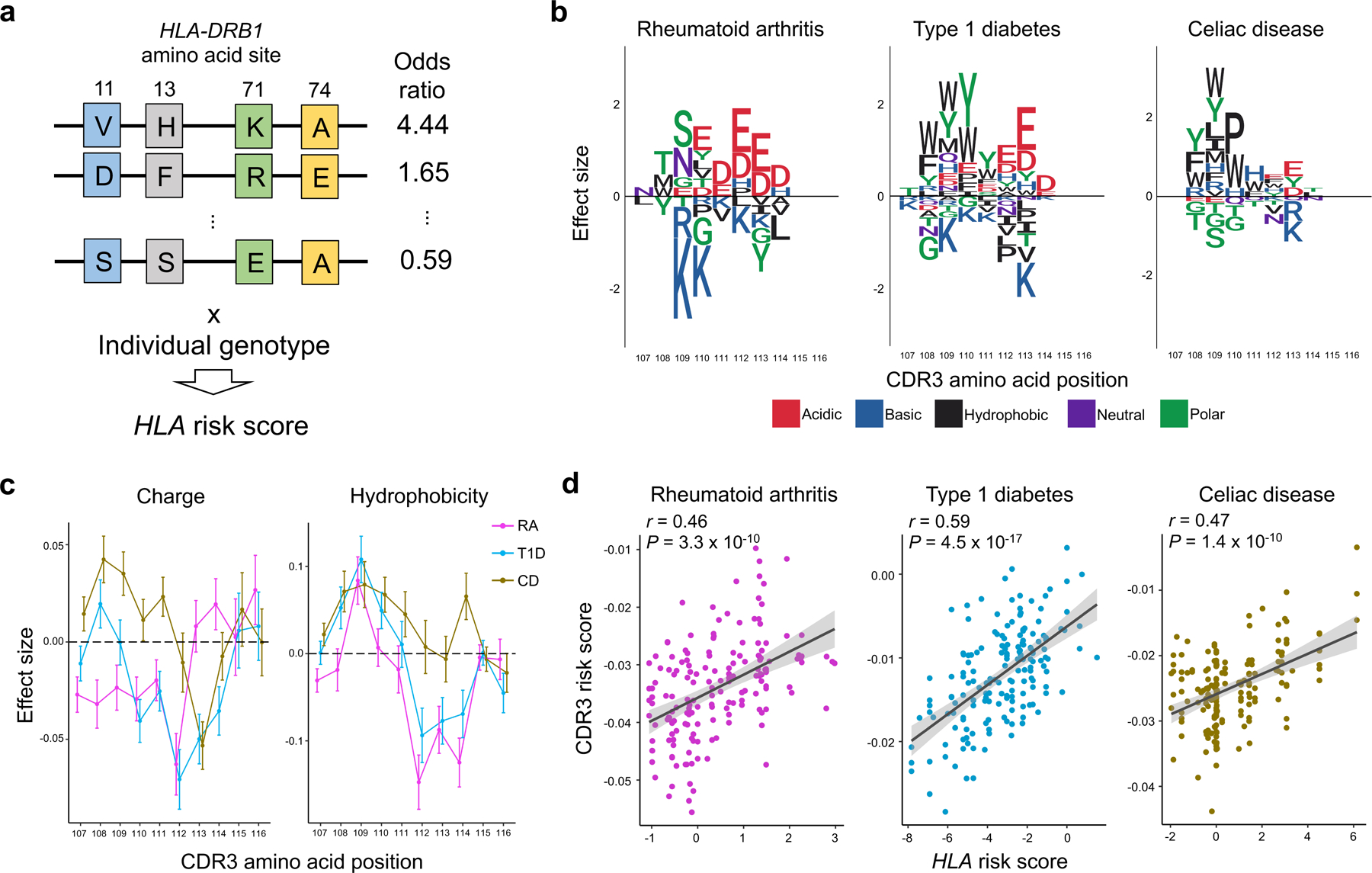
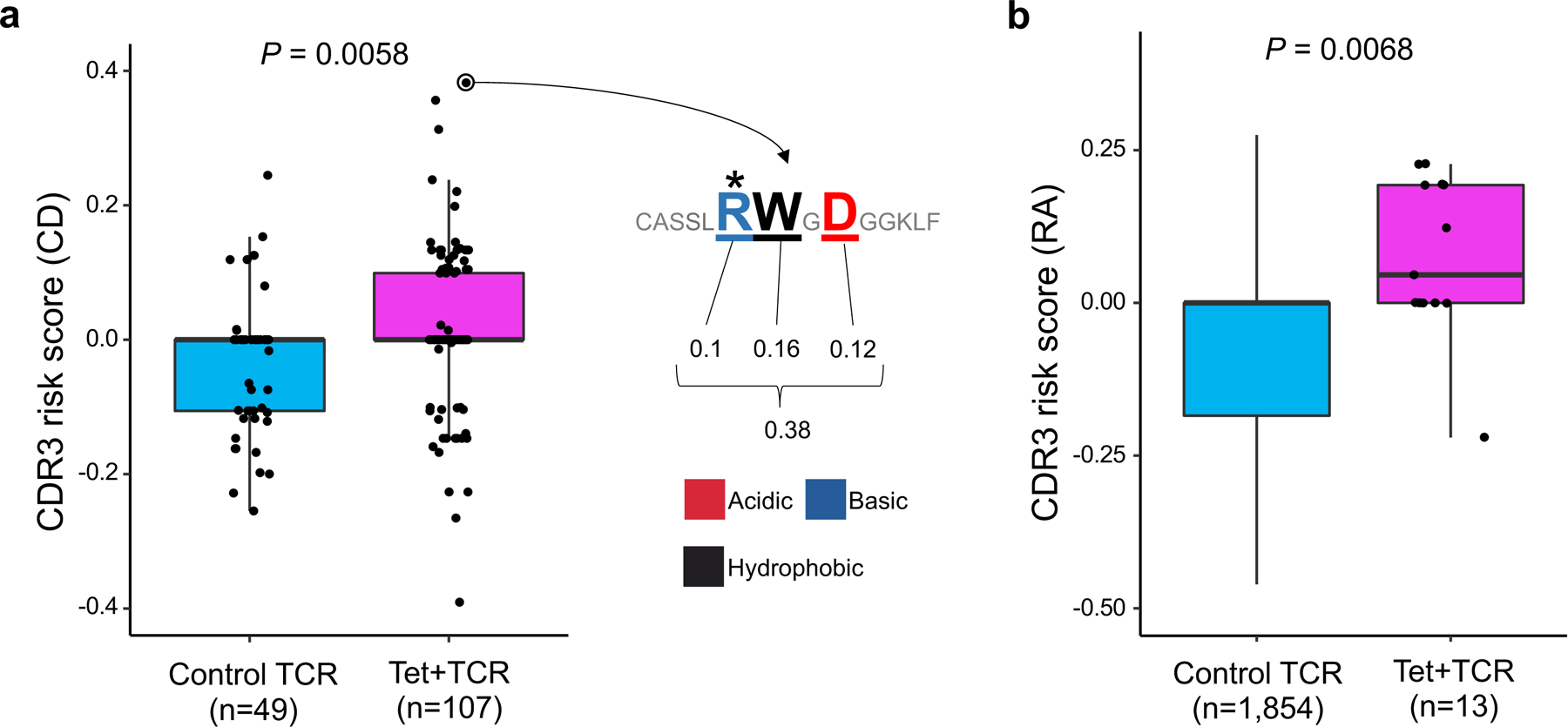
Polymorphisms in the human leukocyte antigen (HLA) genes strongly influence autoimmune disease risk. HLA risk alleles may influence thymic selection to increase the frequency of T cell receptors (TCRs) reactive to autoantigens (central hypothesis). However, research in human autoimmunity has provided little evidence supporting the central hypothesis. Here we investigated the influence of HLA alleles on TCR composition at the highly diverse complementarity determining region 3 (CDR3), which confers antigen recognition. We observed unexpectedly strong HLA-CDR3 associations. The strongest association was found at HLA-DRB1 amino acid position 13, the position that mediates genetic risk for multiple autoimmune diseases. We identified multiple CDR3 amino acid features enriched by HLA risk alleles. Moreover, the CDR3 features promoted by the HLA risk alleles are more enriched in candidate pathogenic TCRs than control TCRs (for example, citrullinated epitope-specific TCRs in patients with rheumatoid arthritis). Together, these results provide genetic evidence supporting the central hypothesis.
© 2022. The Author(s), under exclusive licence to Springer Nature America, Inc.
Conflict of interest statement
COMPETING INTERESTS
The authors declare no competing interests.
Figures
Comment in
-
HLA autoimmunity risk alleles influence T cell receptor sequences.Nat Rev Rheumatol. 2022 Jun;18(6):305. doi: 10.1038/s41584-022-00785-5. Nat Rev Rheumatol. 2022. PMID: 35440763 No abstract available.
Similar articles
-
Mycobacterium tuberculosis peptide E7/HLA-DRB1 tetramers with different HLA-DR alleles bound CD4+ T cells might share identical CDR3 region.Sci Rep. 2018 Jul 2;8(1):9903. doi: 10.1038/s41598-018-28344-7. Sci Rep. 2018. PMID: 29967390 Free PMC article.
-
Collagen Specific T-Cell Repertoire and HLA-DR Alleles: Biomarkers of Active Refractory Rheumatoid Arthritis.EBioMedicine. 2015 Nov 17;2(12):2037-45. doi: 10.1016/j.ebiom.2015.11.019. eCollection 2015 Dec. EBioMedicine. 2015. PMID: 26844284 Free PMC article.
-
T-cell expansions with conserved T-cell receptor beta chain motifs in the peripheral blood of HLA-DRB1*0401 positive patients with necrotizing vasculitis.Blood. 1998 Nov 15;92(10):3737-44. Blood. 1998. PMID: 9808568
-
Associations of human leukocyte antigens with autoimmune diseases: challenges in identifying the mechanism.J Hum Genet. 2015 Nov;60(11):697-702. doi: 10.1038/jhg.2015.100. Epub 2015 Aug 20. J Hum Genet. 2015. PMID: 26290149 Review.
-
Genetics of rheumatoid arthritis.Semin Immunopathol. 2022 Jan;44(1):47-62. doi: 10.1007/s00281-022-00912-0. Epub 2022 Jan 27. Semin Immunopathol. 2022. PMID: 35088123 Free PMC article. Review.
Cited by
-
Immunopeptidomics for autoimmunity: unlocking the chamber of immune secrets.NPJ Syst Biol Appl. 2025 Jan 17;11(1):10. doi: 10.1038/s41540-024-00482-x. NPJ Syst Biol Appl. 2025. PMID: 39833247 Free PMC article. Review.
-
Deciphering state-dependent immune features from multi-layer omics data at single-cell resolution.Nat Genet. 2025 Aug;57(8):1905-1921. doi: 10.1038/s41588-025-02266-3. Epub 2025 Jul 28. Nat Genet. 2025. PMID: 40721531 Free PMC article.
-
Mapping the dynamic genetic regulatory architecture of HLA genes at single-cell resolution.medRxiv [Preprint]. 2023 Mar 20:2023.03.14.23287257. doi: 10.1101/2023.03.14.23287257. medRxiv. 2023. Update in: Nat Genet. 2023 Dec;55(12):2255-2268. doi: 10.1038/s41588-023-01586-6. PMID: 36993194 Free PMC article. Updated. Preprint.
-
The genetic basis of autoimmunity seen through the lens of T cell functional traits.Nat Commun. 2024 Feb 8;15(1):1204. doi: 10.1038/s41467-024-45170-w. Nat Commun. 2024. PMID: 38331990 Free PMC article. Review.
-
Phage display sequencing reveals that genetic, environmental, and intrinsic factors influence variation of human antibody epitope repertoire.Immunity. 2023 Jun 13;56(6):1376-1392.e8. doi: 10.1016/j.immuni.2023.04.003. Epub 2023 May 9. Immunity. 2023. PMID: 37164013 Free PMC article.
References
METHODS-ONLY REFERENCES
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
