

Statistical and machine learning methods for spatially resolved transcriptomics data analysis
- PMID: 35337374
- PMCID: PMC8951701
- DOI: 10.1186/s13059-022-02653-7
Statistical and machine learning methods for spatially resolved transcriptomics data analysis
Abstract
The recent advancement in spatial transcriptomics technology has enabled multiplexed profiling of cellular transcriptomes and spatial locations. As the capacity and efficiency of the experimental technologies continue to improve, there is an emerging need for the development of analytical approaches. Furthermore, with the continuous evolution of sequencing protocols, the underlying assumptions of current analytical methods need to be re-evaluated and adjusted to harness the increasing data complexity. To motivate and aid future model development, we herein review the recent development of statistical and machine learning methods in spatial transcriptomics, summarize useful resources, and highlight the challenges and opportunities ahead.
© 2022. The Author(s).
Conflict of interest statement
The authors declare that they have no competing interests.
Figures
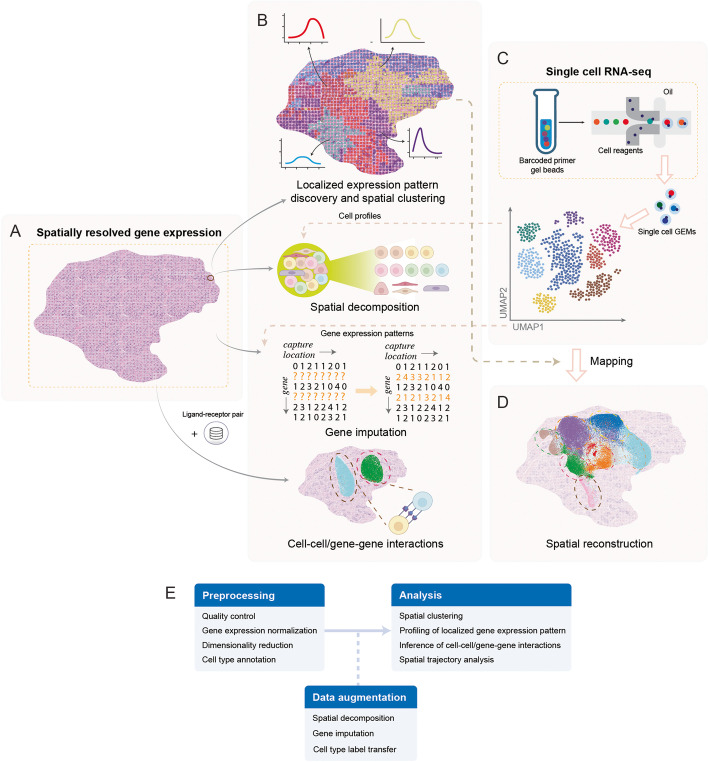
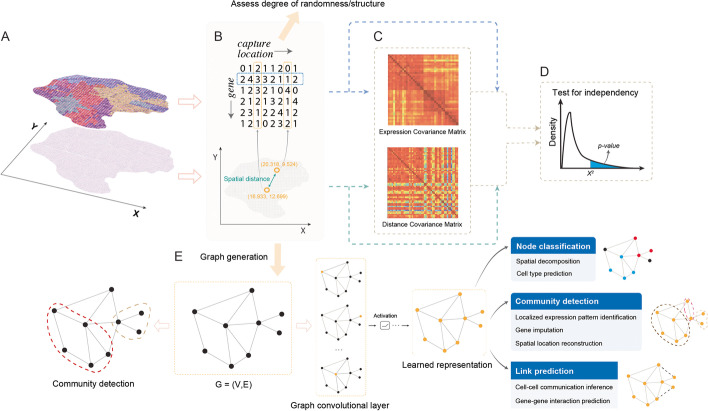
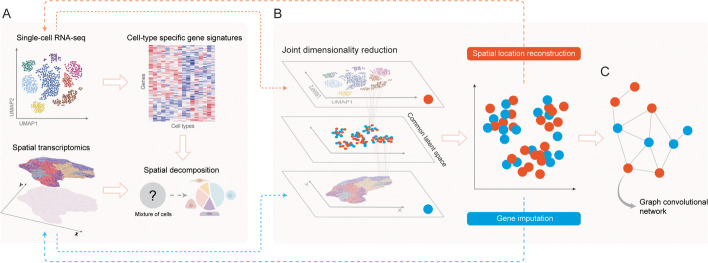
References
-
- Stahl PL, Salmen F, Vickovic S, Lundmark A, Navarro JF, Magnusson J, Giacomello S, Asp M, Westholm JO, Huss M, et al. Visualization and analysis of gene expression in tissue sections by spatial transcriptomics. Science. 2016;353:78–82. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
