

Metagenomic strain detection with SameStr: identification of a persisting core gut microbiota transferable by fecal transplantation
- PMID: 35337386
- PMCID: PMC8951724
- DOI: 10.1186/s40168-022-01251-w
Metagenomic strain detection with SameStr: identification of a persisting core gut microbiota transferable by fecal transplantation
Abstract
Background: The understanding of how microbiomes assemble, function, and evolve requires metagenomic tools that can resolve microbiota compositions at the strain level. However, the identification and tracking of microbial strains in fecal metagenomes is challenging and available tools variably classify subspecies lineages, which affects their applicability to infer microbial persistence and transfer.
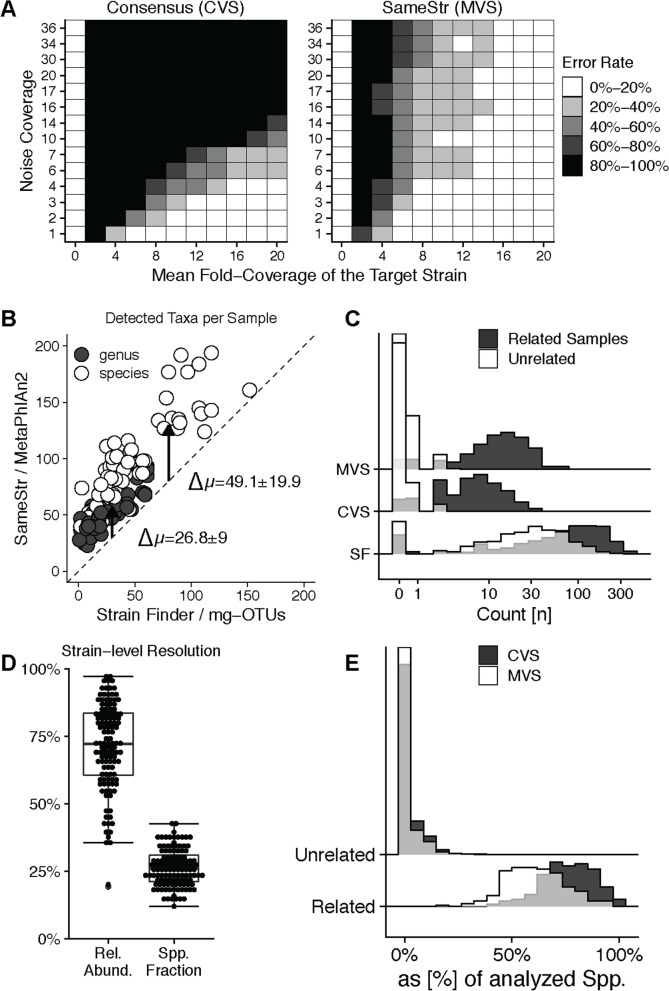
Results: We introduce SameStr, a bioinformatic tool that identifies shared strains in metagenomes by determining single-nucleotide variants (SNV) in species-specific marker genes, which are compared based on a maximum variant profile similarity. We validated SameStr on mock strain populations, available human fecal metagenomes from healthy individuals and newly generated data from recurrent Clostridioides difficile infection (rCDI) patients treated with fecal microbiota transplantation (FMT). SameStr demonstrated enhanced sensitivity to detect shared dominant and subdominant strains in related samples (where strain persistence or transfer would be expected) when compared to other tools, while being robust against false-positive shared strain calls between unrelated samples (where neither strain persistence nor transfer would be expected). We applied SameStr to identify strains that are stably maintained in fecal microbiomes of healthy adults over time (strain persistence) and that successfully engraft in rCDI patients after FMT (strain engraftment). Taxonomy-dependent strain persistence and engraftment frequencies were positively correlated, indicating that a specific core microbiota of intestinal species is adapted to be competitive both in healthy microbiomes and during post-FMT microbiome assembly. We explored other use cases for strain-level microbiota profiling, as a metagenomics quality control measure and to identify individuals based on the persisting core gut microbiota.
Conclusion: SameStr provides for a robust identification of shared strains in metagenomic sequence data with sufficient specificity and sensitivity to examine strain persistence, transfer, and engraftment in human fecal microbiomes. Our findings identify a persisting healthy adult core gut microbiota, which should be further studied to shed light on microbiota contributions to chronic diseases. Video abstract.
© 2022. The Author(s).
Conflict of interest statement
The authors declare that they have no competing interests.
Figures




References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
