

Comprehensive characterization of PTEN mutational profile in a series of 34,129 colorectal cancers
- PMID: 35338148
- PMCID: PMC8956741
- DOI: 10.1038/s41467-022-29227-2
Comprehensive characterization of PTEN mutational profile in a series of 34,129 colorectal cancers
Abstract
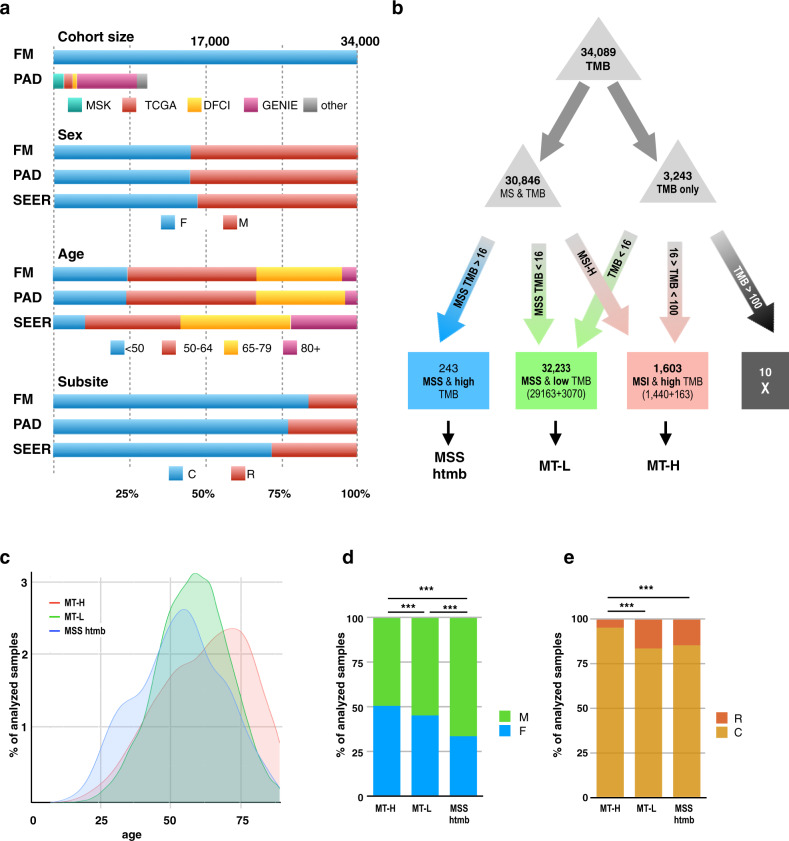
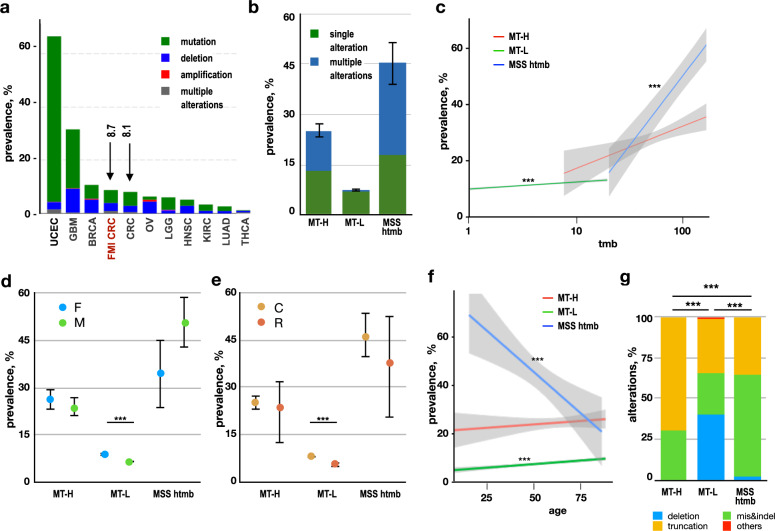
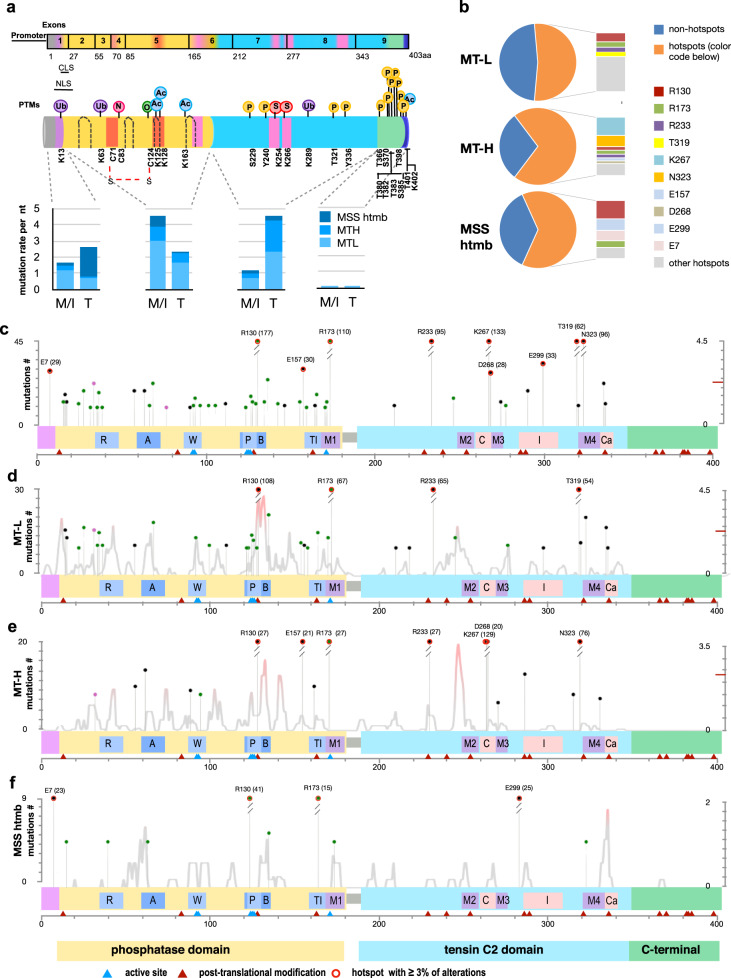
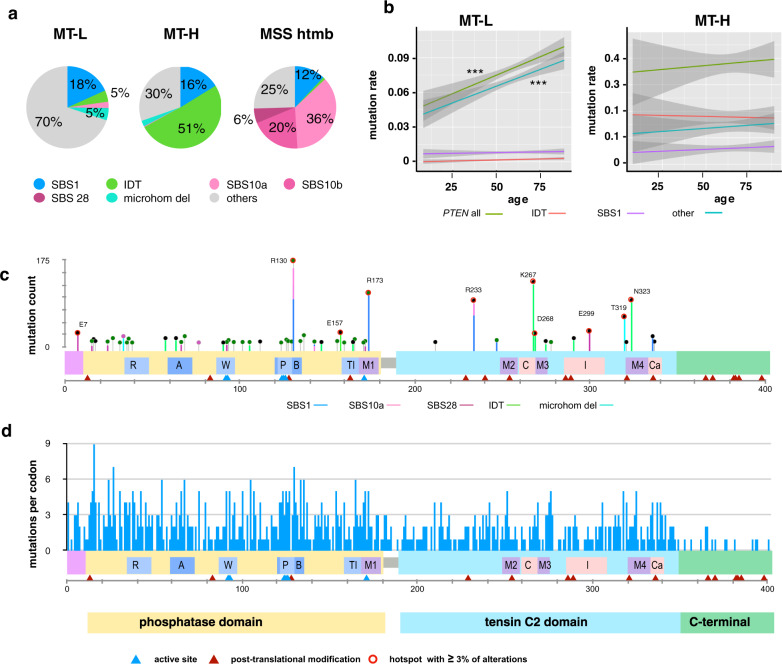
Loss of expression or activity of the tumor suppressor PTEN acts similarly to an activating mutation in the oncogene PIK3CA in elevating intracellular levels of phosphatidylinositol (3,4,5)-trisphosphate (PIP3), inducing signaling by AKT and other pro-tumorigenic signaling proteins. Here, we analyze sequence data for 34,129 colorectal cancer (CRC) patients, capturing 3,434 PTEN mutations. We identify specific patterns of PTEN mutation associated with microsatellite stability/instability (MSS/MSI), tumor mutational burden (TMB), patient age, and tumor location. Within groups separated by MSS/MSI status, this identifies distinct profiles of nucleotide hotspots, and suggests differing profiles of protein-damaging effects of mutations. Moreover, discrete categories of PTEN mutations display non-identical patterns of co-occurrence with mutations in other genes important in CRC pathogenesis, including KRAS, APC, TP53, and PIK3CA. These data provide context for clinical targeting of proteins upstream and downstream of PTEN in distinct CRC cohorts.
© 2022. The Author(s).
Conflict of interest statement
J.N. and G.F. are employed by FMI and own stock in Roche. The remaining authors declare no competing interests.
Figures
Similar articles
-
DNA sequence profiles of the colorectal cancer critical gene set KRAS-BRAF-PIK3CA-PTEN-TP53 related to age at disease onset.PLoS One. 2010 Nov 12;5(11):e13978. doi: 10.1371/journal.pone.0013978. PLoS One. 2010. PMID: 21103049 Free PMC article.
-
Mutational profiling of colorectal cancers with microsatellite instability.Oncotarget. 2015 Dec 8;6(39):42334-44. doi: 10.18632/oncotarget.5997. Oncotarget. 2015. PMID: 26517354 Free PMC article.
-
Molecular alterations of Ras-Raf-mitogen-activated protein kinase and phosphatidylinositol 3-kinase-Akt signaling pathways in colorectal cancers from a tertiary hospital at Kuala Lumpur, Malaysia.APMIS. 2013 Oct;121(10):954-66. doi: 10.1111/apm.12152. Epub 2013 Aug 29. APMIS. 2013. PMID: 23992303
-
Recommendations from the EGAPP Working Group: can testing of tumor tissue for mutations in EGFR pathway downstream effector genes in patients with metastatic colorectal cancer improve health outcomes by guiding decisions regarding anti-EGFR therapy?Genet Med. 2013 Jul;15(7):517-27. doi: 10.1038/gim.2012.184. Epub 2013 Feb 21. Genet Med. 2013. PMID: 23429431
-
PIK3CA mutation and clinicopathological features of colorectal cancer: a systematic review and Meta-Analysis.Acta Oncol. 2020 Jan;59(1):66-74. doi: 10.1080/0284186X.2019.1664764. Epub 2019 Sep 23. Acta Oncol. 2020. PMID: 31545109
Cited by
-
Source, co-occurrence, and prognostic value of PTEN mutations or loss in colorectal cancer.NPJ Genom Med. 2023 Nov 24;8(1):40. doi: 10.1038/s41525-023-00384-7. NPJ Genom Med. 2023. PMID: 38001126 Free PMC article.
-
MDM2 inhibition is synthetic lethal with PTEN loss in colorectal cancer cells via the p53-dependent mechanism.Int J Biol Sci. 2023 Jul 9;19(11):3544-3557. doi: 10.7150/ijbs.82566. eCollection 2023. Int J Biol Sci. 2023. PMID: 37496993 Free PMC article.
-
Cell-type specific defects in PTEN-mutant cortical organoids converge on abnormal circuit activity.Hum Mol Genet. 2023 Sep 5;32(18):2773-2786. doi: 10.1093/hmg/ddad107. Hum Mol Genet. 2023. PMID: 37384417 Free PMC article.
-
Exploring Co-occurring POLE Exonuclease and Non-exonuclease Domain Mutations and Their Impact on Tumor Mutagenicity.Cancer Res Commun. 2024 Jan 26;4(1):213-225. doi: 10.1158/2767-9764.CRC-23-0312. Epub 2024 Jan 8. Cancer Res Commun. 2024. PMID: 38282550 Free PMC article.
-
Pan-cancer genomic analysis shows hemizygous PTEN loss tumors are associated with immune evasion and poor outcome.Sci Rep. 2023 Mar 28;13(1):5049. doi: 10.1038/s41598-023-31759-6. Sci Rep. 2023. PMID: 36977733 Free PMC article.
References
-
- Siegel RL, et al. Colorectal cancer statistics. CA Cancer J. Clin. 2021;71:7–33. - PubMed
-
- Dienstmann R, et al. Consensus molecular subtypes and the evolution of precision medicine in colorectal cancer. Nat. Rev. Cancer. 2017;17:79–92. - PubMed
-
- Benedix F, et al. Comparison of 17,641 patients with right- and left-sided colon cancer: differences in epidemiology, perioperative course, histology, and survival. Dis. Colon Rectum. 2010;53:57–64. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
