

Simulations of molecular photodynamics in long timescales
- PMID: 35341303
- PMCID: PMC8958277
- DOI: 10.1098/rsta.2020.0382
Simulations of molecular photodynamics in long timescales
Abstract
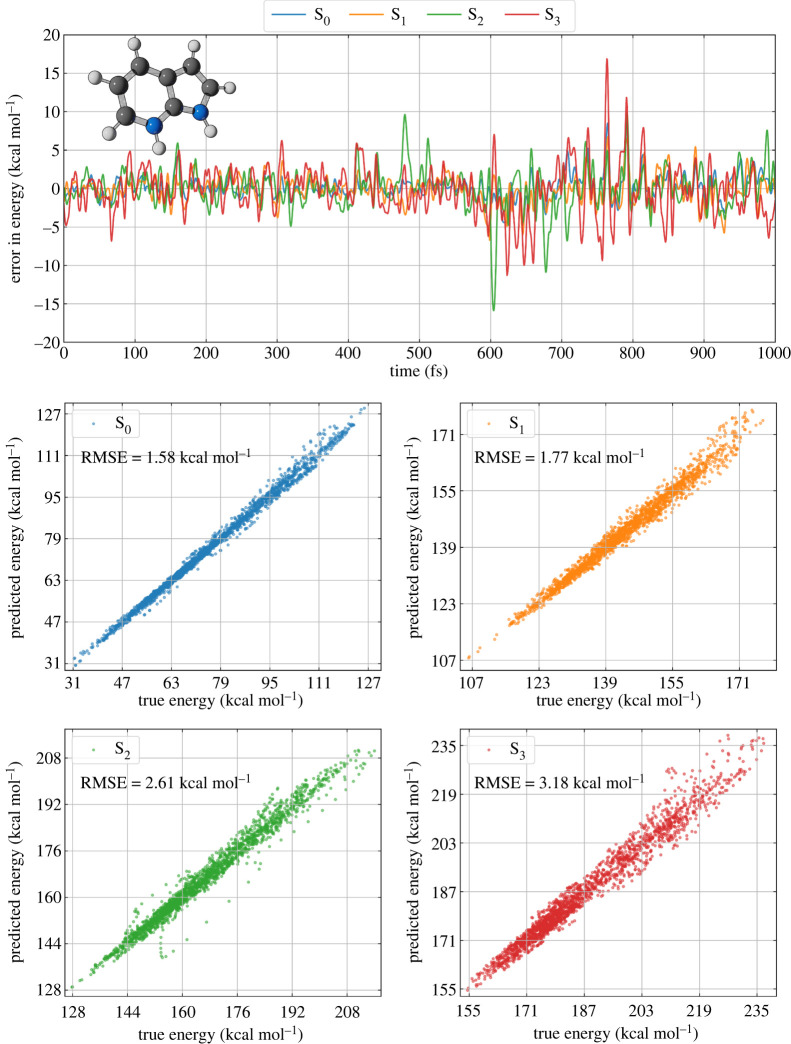
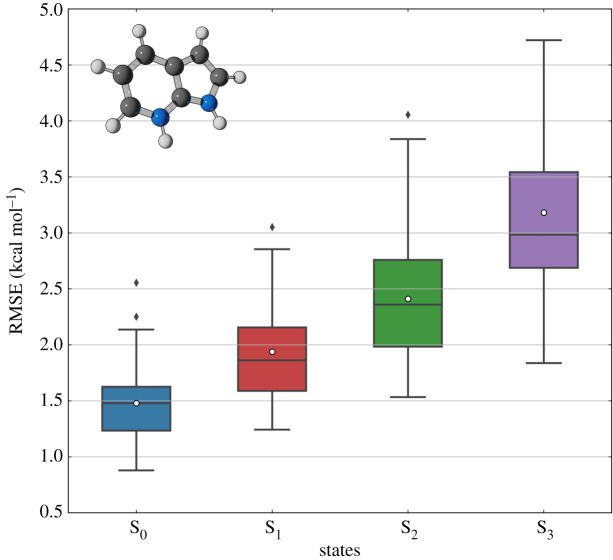
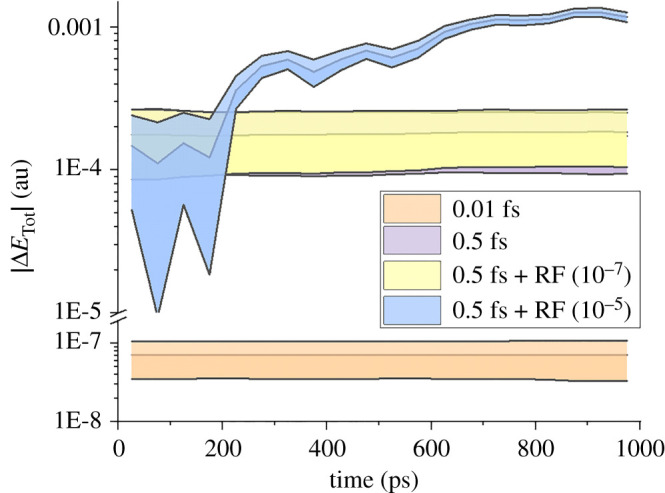
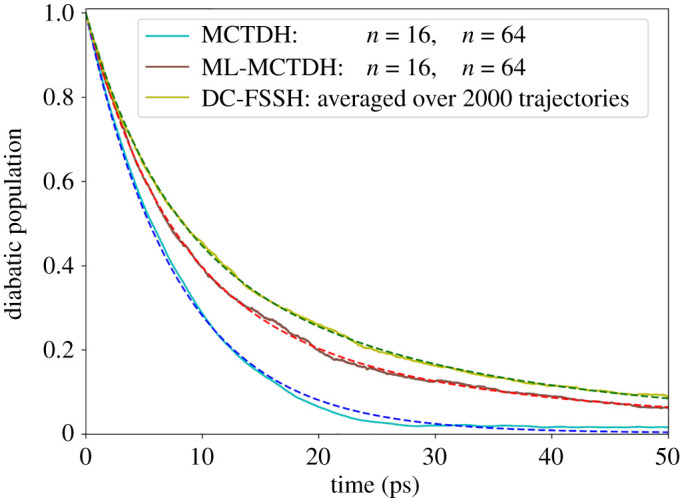
Nonadiabatic dynamics simulations in the long timescale (much longer than 10 ps) are the next challenge in computational photochemistry. This paper delimits the scope of what we expect from methods to run such simulations: they should work in full nuclear dimensionality, be general enough to tackle any type of molecule and not require unrealistic computational resources. We examine the main methodological challenges we should venture to advance the field, including the computational costs of the electronic structure calculations, stability of the integration methods, accuracy of the nonadiabatic dynamics algorithms and software optimization. Based on simulations designed to shed light on each of these issues, we show how machine learning may be a crucial element for long time-scale dynamics, either as a surrogate for electronic structure calculations or aiding the parameterization of model Hamiltonians. We show that conventional methods for integrating classical equations should be adequate to extended simulations up to 1 ns and that surface hopping agrees semiquantitatively with wave packet propagation in the weak-coupling regime. We also describe our optimization of the Newton-X program to reduce computational overheads in data processing and storage. This article is part of the theme issue 'Chemistry without the Born-Oppenheimer approximation'.
Keywords: computational chemistry; dynamics simulations; excited states; nonadiabatic phenomena; photochemistry; theoretical chemistry.
Conflict of interest statement
The authors declare that they have no competing interests.
Figures
References
-
- Vacher M, Mendive-Tapia D, Bearpark MJ, Robb MA. 2014. The second-order Ehrenfest method. Theor. Chem. Acc. 133, 1505. (10.1007/s00214-014-1505-6) - DOI
LinkOut - more resources
Full Text Sources
Miscellaneous