

Phenotypic and mutational spectrum of ROR2-related Robinow syndrome
- PMID: 35344616
- PMCID: PMC9177636
- DOI: 10.1002/humu.24375
Phenotypic and mutational spectrum of ROR2-related Robinow syndrome
Abstract
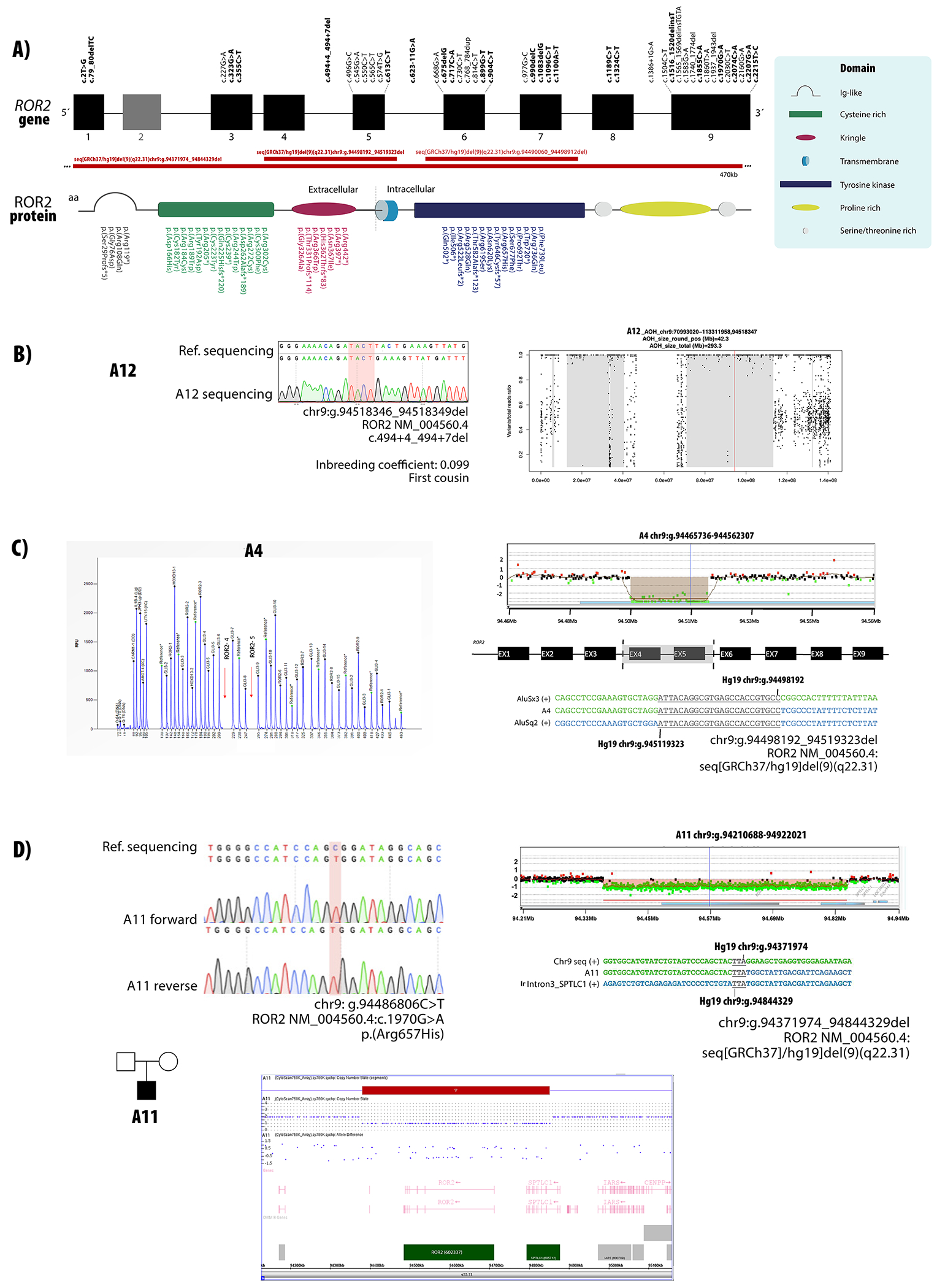
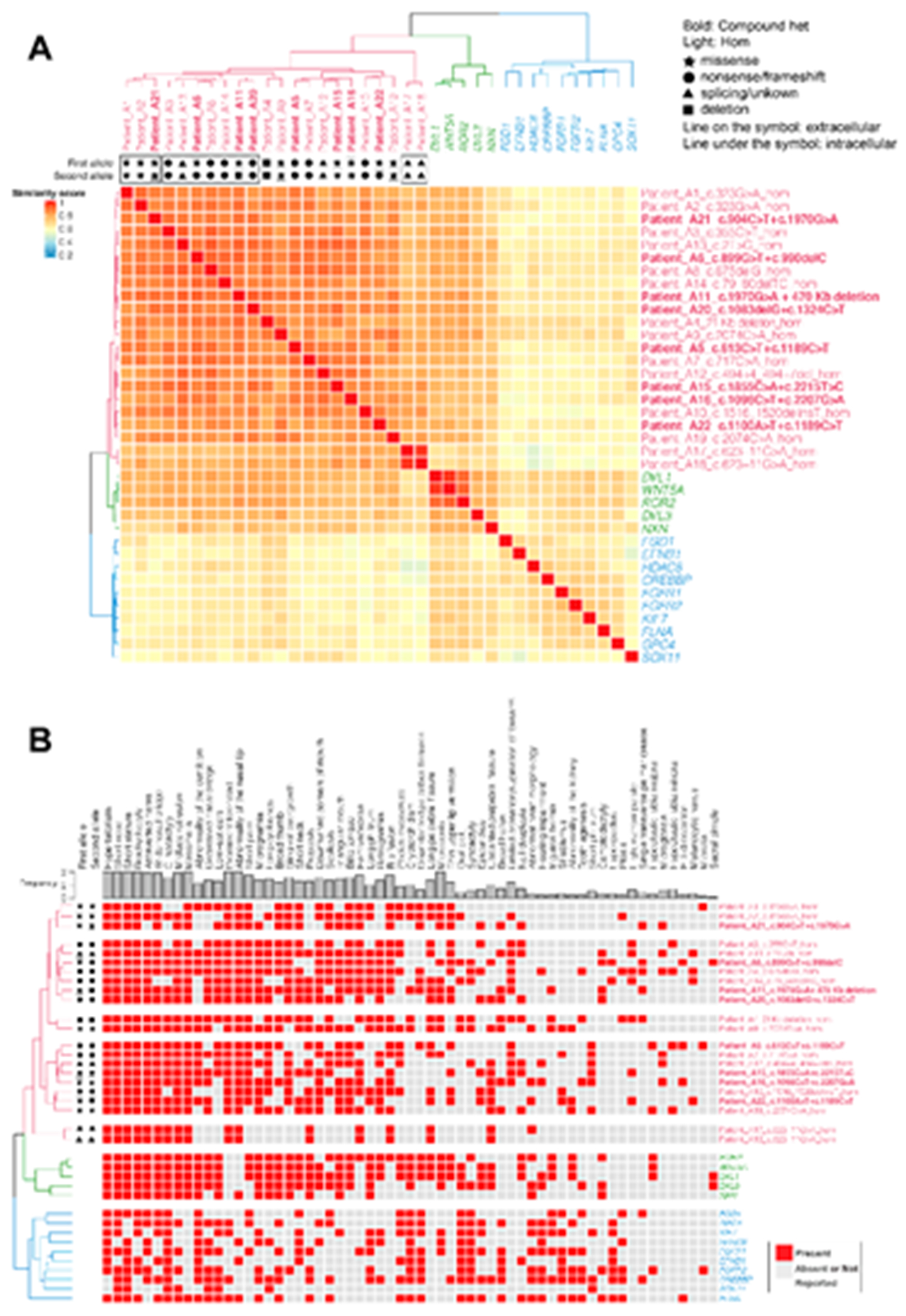
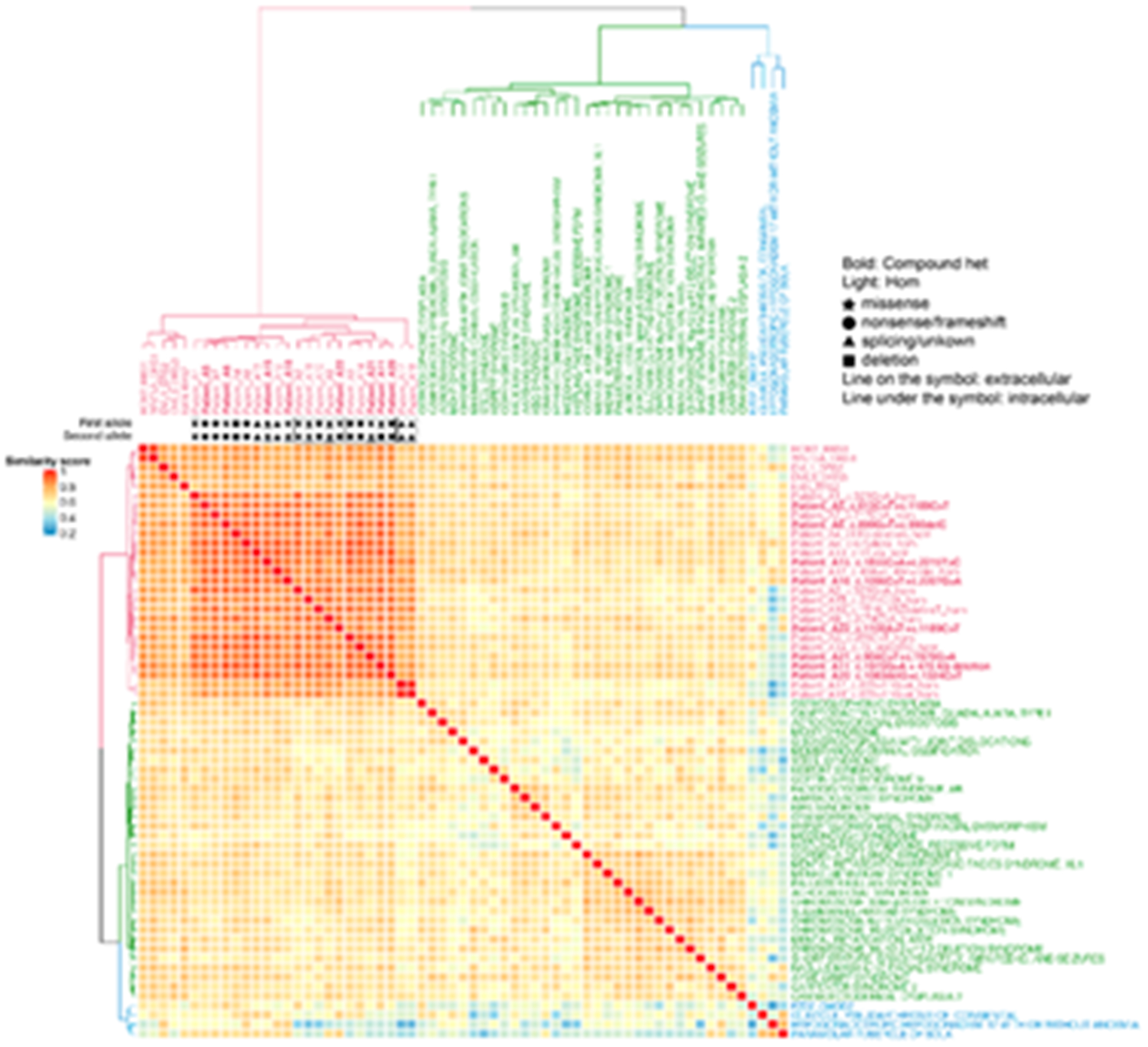
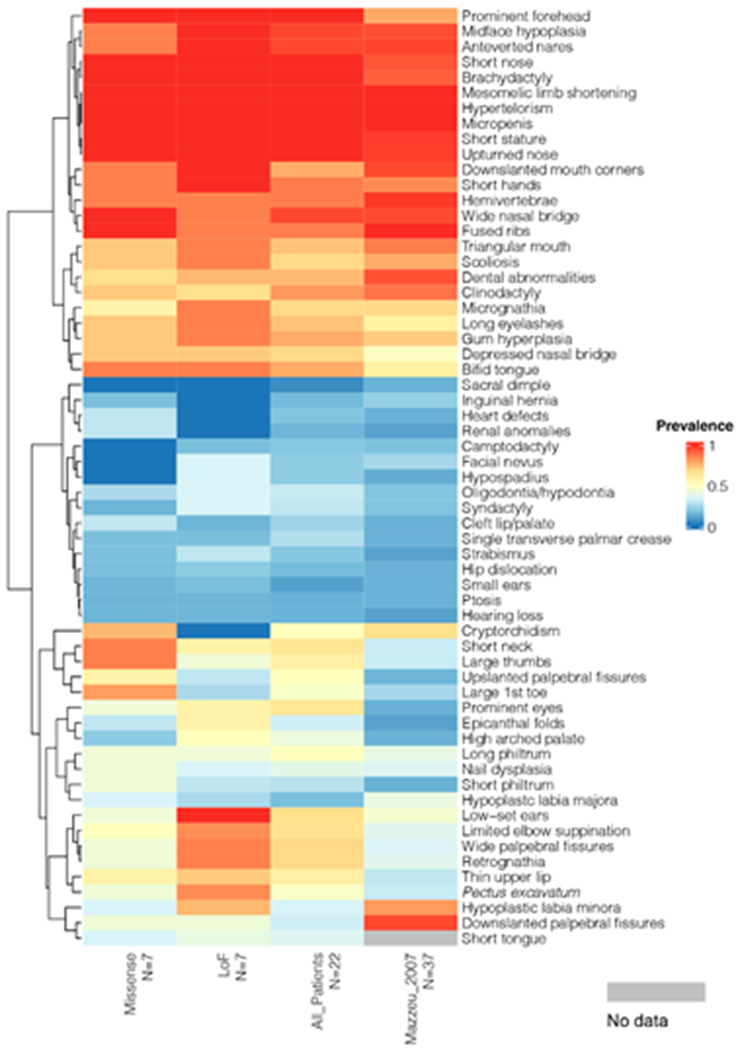
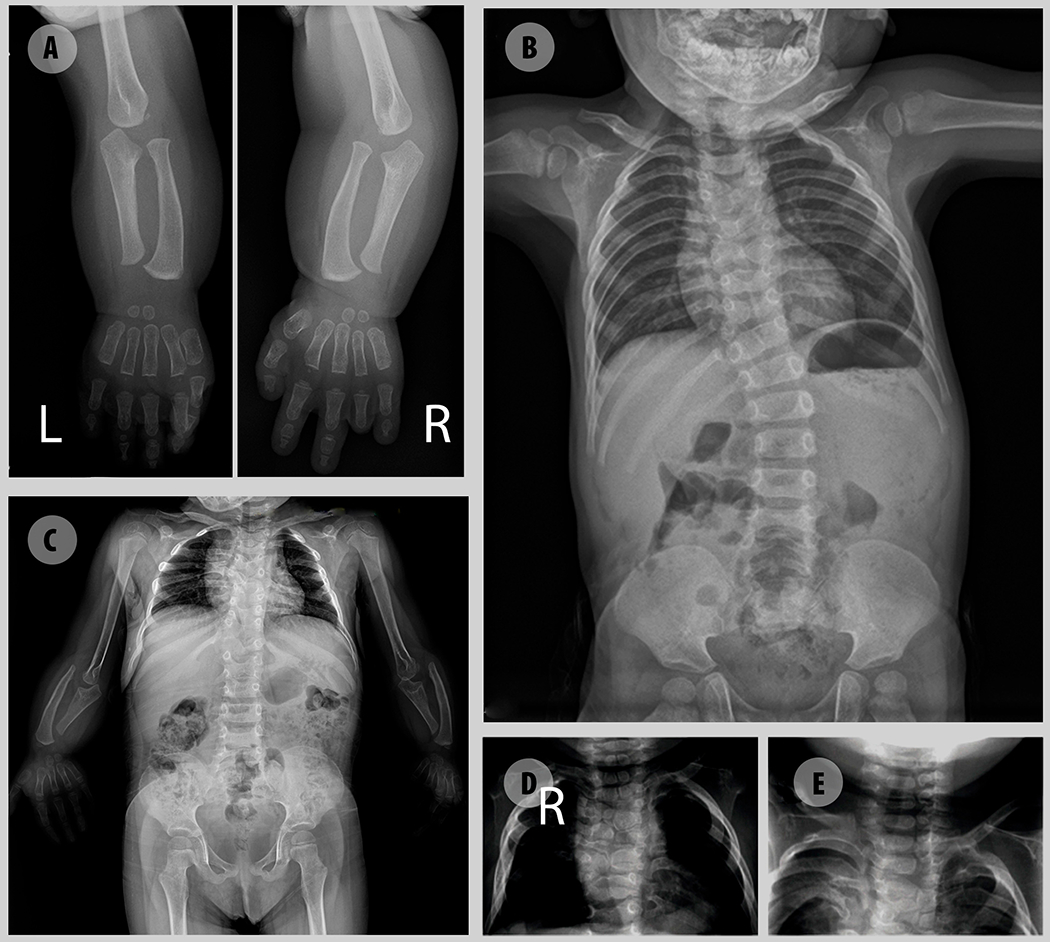
Robinow syndrome is characterized by a triad of craniofacial dysmorphisms, disproportionate-limb short stature, and genital hypoplasia. A significant degree of phenotypic variability seems to correlate with different genes/loci. Disturbances of the noncanonical WNT-pathway have been identified as the main cause of the syndrome. Biallelic variants in ROR2 cause an autosomal recessive form of the syndrome with distinctive skeletal findings. Twenty-two patients with a clinical diagnosis of autosomal recessive Robinow syndrome were screened for variants in ROR2 using multiple molecular approaches. We identified 25 putatively pathogenic ROR2 variants, 16 novel, including single nucleotide variants and exonic deletions. Detailed phenotypic analyses revealed that all subjects presented with a prominent forehead, hypertelorism, short nose, abnormality of the nasal tip, brachydactyly, mesomelic limb shortening, short stature, and genital hypoplasia in male patients. A total of 19 clinical features were present in more than 75% of the subjects, thus pointing to an overall uniformity of the phenotype. Disease-causing variants in ROR2, contribute to a clinically recognizable autosomal recessive trait phenotype with multiple skeletal defects. A comprehensive quantitative clinical evaluation of this cohort delineated the phenotypic spectrum of ROR2-related Robinow syndrome. The identification of exonic deletion variant alleles further supports the contention of a loss-of-function mechanism in the etiology of the syndrome.
Keywords: HPO terms; WNT pathway; chromosome microarray analysis; craniofacial morphology; exonic deletion; next-generation sequencing; quantitative phenotyping cluster heatmap; skeletal dysplasia.
© 2022 Wiley Periodicals LLC.
Conflict of interest statement
Conflict of interests:
Baylor College of Medicine (BCM) and Miraca Holdings have formed a joint venture with shared ownership and governance of the Baylor Genetics (BG), which performs clinical microarray analysis and clinical exome sequencing and whole genome sequencing. J.R.L. serves on the Scientific Advisory Board of the BG. J.R.L. has stock ownership in 23andMe, is a paid consultant for Regeneron Genetics Center, and is a coinventor on multiple United States and European patents related to molecular diagnostics for inherited neuropathies, eye diseases, genomic disorders and bacterial genomic fingerprinting. The Department of Molecular and Human Genetics at Baylor College of Medicine derives revenue from molecular genetics and clinical genomics testing offered at BG. The other authors declare no competing financial interests.
Figures

References
-
- Aglan M, Amr K, Ismail S, Ashour A, Otaify GA, Mehrez MA, Aboul-Ezz EH, El-Ruby M, Mazen I, Abdel-Hamid MS, & Temtamy SA (2015). Clinical and molecular characterization of seven Egyptian families with autosomal recessive robinow syndrome: Identification of four novel ROR2 gene mutations. American journal of medical genetics. Part A, 167A(12), 3054–3061. 10.1002/ajmg.a.37287 - DOI - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
