

L-alanine supplementation in Pompe disease (IOPD): a potential therapeutic implementation for patients on ERT? A case report
- PMID: 35346323
- PMCID: PMC8962230
- DOI: 10.1186/s13052-022-01249-y
L-alanine supplementation in Pompe disease (IOPD): a potential therapeutic implementation for patients on ERT? A case report
Abstract
Background: Pompe disease (PD) is a disorder of glycogen metabolism conditioning a progressive and life conditioning myopathy. Enzyme replacement therapy (ERT) is currently the best treatment option for PD, but is not resolutive. While other potential therapeutic approaches have been reported before, these have never been tried as co- treatments. L-alanine oral supplementation (LAOS) has been proven to reduce muscle breakdown: we hereby report the first case of supplementation on a PD patient on ERT.
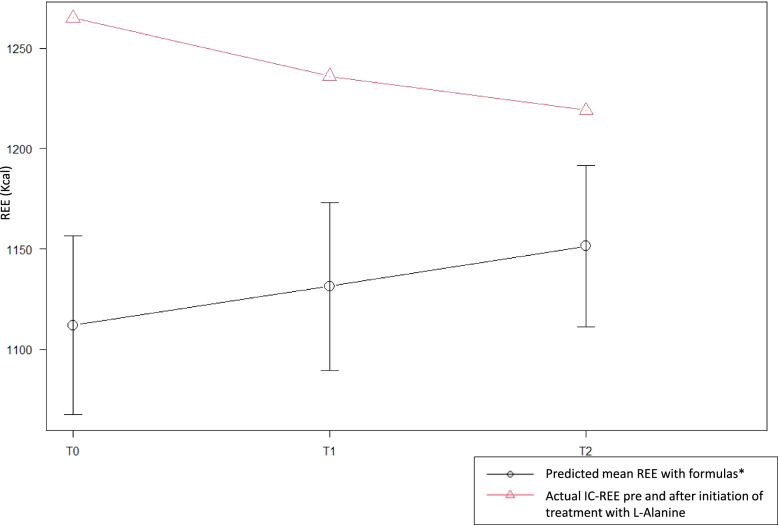
Case presentation: F. is a 9 y.o. infantile onset Pompe Disease (IOPD) girl ERT-treated since age 1 developing a progressive myopathy. We started her on LAOS and performed assessments at baseline, 6 and 9 months. At baseline, F.'s weight, height and BMI were within normal ranges, while body composition showed low fat mass -FM and high resting energy expenditure-REE levels. After LAOS, a progressive FM increase and REE reduction could be observed both at 6 and 9 months.
Conclusions: ERT is not curative for PD patients thus additional treatments could be considered to improve outcomes. Our patient showed physical signs of inability to accumulate energy when exclusively on ERT, while FM increase and REE reduction occurred when supplemented with LAOS, likely reflecting anabolic pathways' implementation. This is the first case reporting potential LAOS benefits in PD-on ERT patients. Longitudinal case control studies are yet needed to evaluate possible efficacy of combined LAOS And ERT treatment in PD patients.
Keywords: Body composition; Enzyme replacement therapy; L-alanine; Myopathy; Pompe Disease.
© 2022. The Author(s).
Conflict of interest statement
The authors declare that they have no competing interests.
Figures
Similar articles
-
Long term clinical history of an Italian cohort of infantile onset Pompe disease treated with enzyme replacement therapy.Orphanet J Rare Dis. 2018 Feb 8;13(1):32. doi: 10.1186/s13023-018-0771-0. Orphanet J Rare Dis. 2018. PMID: 29422078 Free PMC article.
-
Hearing characteristics of infantile-onset Pompe disease after early enzyme-replacement therapy.Orphanet J Rare Dis. 2021 Aug 5;16(1):348. doi: 10.1186/s13023-021-01817-1. Orphanet J Rare Dis. 2021. PMID: 34353347 Free PMC article.
-
Expert Group Consensus on early diagnosis and management of infantile-onset pompe disease in the Gulf Region.Orphanet J Rare Dis. 2022 Oct 27;17(1):388. doi: 10.1186/s13023-022-02545-w. Orphanet J Rare Dis. 2022. PMID: 36303251 Free PMC article. Review.
-
Effect of alglucosidase alfa dosage on survival and walking ability in patients with classic infantile Pompe disease: a multicentre observational cohort study from the European Pompe Consortium.Lancet Child Adolesc Health. 2022 Jan;6(1):28-37. doi: 10.1016/S2352-4642(21)00308-4. Epub 2021 Nov 22. Lancet Child Adolesc Health. 2022. PMID: 34822769
-
Gene Therapy for Pompe Disease: The Time is now.Hum Gene Ther. 2019 Oct;30(10):1245-1262. doi: 10.1089/hum.2019.109. Epub 2019 Sep 9. Hum Gene Ther. 2019. PMID: 31298581 Review.
Cited by
-
Glycogen storage diseases: An update.World J Gastroenterol. 2023 Jul 7;29(25):3932-3963. doi: 10.3748/wjg.v29.i25.3932. World J Gastroenterol. 2023. PMID: 37476587 Free PMC article. Review.
-
Nutritional management of glycogen storage disease type III: a case report and a critical appraisal of the literature.Front Nutr. 2023 May 11;10:1178348. doi: 10.3389/fnut.2023.1178348. eCollection 2023. Front Nutr. 2023. PMID: 37252245 Free PMC article.
-
Single Amino Acid Supplementation in Inherited Metabolic Disorders: An Evidence-Based Review of Interventions.Genes (Basel). 2025 Apr 27;16(5):502. doi: 10.3390/genes16050502. Genes (Basel). 2025. PMID: 40428324 Free PMC article. Review.
-
An Assessment of Dietary Intake, Feeding Practices, Growth, and Swallowing Function in Young Children with Late-Onset Pompe Disease: A Framework for Developing Nutrition Guidelines.Nutrients. 2025 Jun 1;17(11):1909. doi: 10.3390/nu17111909. Nutrients. 2025. PMID: 40507177 Free PMC article.
References
-
- A.J.J. Reuser, R. Hirschhorn, M.A. Kroos, Pompe Disease: Glycogen Storage Disease Type II, Acid α-Glucosidase (Acid Maltase) Deficiency, in: D.L. Valle, S. Antonarakis, A. Ballabio, A.L. Beaudet, G.A. Mitchell, The Online Metabolic and Molecular Bases of Inherited Disease. The McGraw-Hill Companies (2001), 3389–3420 https://ommbid.mhmedical.com/content.aspx?bookid=2709§ionid=225890450).
-
- Amalfitano A, McVie-Wylie AJ, Hu H, et al. Systemic correction of the muscle disorder glycogen storage disease type II after hepatic targeting of a modified adenovirus vector encoding human acid-alpha-glucosidase. Proc Natl Acad Sci USA. 1999;96:8861–8866. doi: 10.1073/pnas.96.16.8861. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
